分子系統学演習 データセットの作成から仮説検定まで
はじめに
本書は、2008年10月の農林交流センターワークショップ「分子系統樹推定法:理論と応用」での講義用に執筆を開始しました。そのため、当初は受講者の復習と落選者の自習のためのものでした。それ以来毎年10月頃にワークショップがあり、それに合わせて加筆・修正を加え、現在の姿になりました。
分子系統樹推定法は、多くの分野で求められている技術となってきましたが、残念なことに未だに確立されたものではなく発展途上です。今回、私の担当する講義では「よく使われている方法」ではなく、(たぶん)「現状では最も良い方法」を提示することにしました。そのため、本書の内容は非常にアグレッシブな内容となっています。本書をお読みの皆さんの中には、既存の論文の中で使われている方法の解説を望まれる方がいらっしゃるかもしれません。おそらくそういう方にも多少は役に立つ情報は載っていると思いますが、本書の目的は「現状では最も良い(と私が勝手に思っている)方法」を提示することにあることを予めご了承下さい。
また、本書はクリエイティブ・コモンズの表示-継承 2.1 日本ライセンスの下で配布することにしました。このライセンスの下では、原著作者の明示を行う限り、利用者は自由に本書を複製・頒布・展示することができます。また、原著作者の明示と本ライセンスまたは互換性のあるライセンスの適用を行う限り、本書を改変した二次著作物の作成・配布も自由に行うことができます。詳しい使用許諾条件を見るには
http://creativecommons.org/licenses/by-sa/2.1/jp/
をチェックするか、クリエイティブ・コモンズに郵便にてお問い合わせください。住所は:171 Second Street, Suite 300, San Francisco, California 94105, USA です。
本書が皆さんの役に立つことができましたら幸いです。この機会を与えて下さった農業環境技術研究所の三中信宏先生と、本書をお読みの皆さんに感謝します。
凡例
本書ではコンピュータに入力するコマンドやその結果を表記する際に以下のように記述しています。
# コメント > command option1 \ option2 \ option3↓ output of command > command option1 option2 option3↓ output of command
上記の例ではcommand option1 option2 option3という全く同じコマンドを2回実行しており、コマンド実行後にoutput of commandがコマンドにより表示されています。ここで、#から改行まではコメントを表しており、入力の必要はありません。行頭の>とそれに続くスペースはコマンドの入力の開始を表しており、↓までがコマンドとオプションの入力内容になります。>とそれに続くスペースはあくまで入力の開始を示すためのものですので、入力しないで下さい。↓は入力の終端を表し、ここでEnterキーを押すことを指示する記号です。↓を入力しないようにして下さい。なお、コマンドとオプションを見やすくするためにコマンドやオプションの途中に改行を意図的に入れることがありますが、そのような改行の直前には\を記してあります。したがって、\が直前にある改行はコマンドの終端や改行入力の指示を意味しません。また、表示環境によってはワードラップ機能により筆者の意図しない改行が入ってしまうことがありますが、これもコマンドの終端や改行入力の指示を意味しませんので注意して下さい。
また、本書では様々なファイルを使用しますが、その内容は以下のように記述しています。
| 1行目の内容 | 2行目の内容
この例では、行頭の|とそれに続くスペースはファイル内の行頭を表しており、ファイル作成の際は入力しないように注意して下さい。これは、ワードラップ機能による筆者の意図しない改行とファイルに入力すべき改行を区別できるようにするためのものです。
第0章 必要なソフトウェアのインストールと環境整備
本書が想定するのは、Windows・Linux・Mac OS Xの3つのOSです。各OSのバージョンはWindowsではXP~7、LinuxではDebian GNU/Linux wheezyかUbuntu 12.04 LTS、Mac OS XではSnow Leopard以降のことしか想定していません。これら以外のOSでは、自力で何とかしていただく必要があります。上記のOSであっても、場合によっては自力で何とかする必要性に直面する可能性はあります。
0.1 Windowsの場合
Jalview、TracerおよびFigTreeの動作にはJava実行環境が必要ですが、Windowsには標準では最新のJava実行環境が備わっていません。そのため
http://java.com/
からJava実行環境を入手してインストールしておく必要があります。
また、Windows環境では、エクスプローラ上で指定したフォルダをカレントフォルダとするコマンドプロンプトを簡単に起動できるようになる「ContextConsole Shell Extension」をインストールしておくことをおすすめします。
http://code.kliu.org/cmdopen/
から入手できます。このソフトをインストールすると、フォルダアイコンの右クリックメニューからコマンドプロンプトを起動できるようになります。なお、「カレントフォルダ」というのは、その時点でプログラムを起動すると作業フォルダとして使われるフォルダのことです。プログラムによってはカレントフォルダを無視して任意のフォルダを作業フォルダとするものもあります。フォルダのことをディレクトリと呼ぶこともありますが意味は同じです。
Windowsでは、標準ではファイル名末尾の拡張子(.fasとか.nexのこと)が表示されません。これはこの先大変不便なので、表示するように変更しておく必要があります。それにはまず、エクスプローラを起動(WinキーとEの同時押しで可能です)し、ツールメニュー内のフォルダ オプションを開きます(Vista/7ではコントロールパネル内にもあります)。すると、表示されるダイアログに表示タブがありますのでそれを選択します。そして、詳細設定ペインの中に登録されている拡張子は表示しないという項目があり、チェックが入っているはずですので、そのチェックを外してOKを押すと、拡張子が表示されるようになります。また、Windows Vista/7に搭載されているユーザーアカウント制御(UAC)という機能は、セキュリティ上重要ではあるのですが、様々なソフトが正常に動作しないようにしてしまう困った機能ですので、もし何か問題があれば無効にして試してみて下さい。ウィルス対策ソフトも誤検出や暴走するものがありますので注意して下さい。
後の操作の際に、「正規表現」を利用した検索・置換が可能なテキストエディタがあると便利です。正規表現とは、「一定のルールに該当する文字列を検索する」ためのそのルールの記述方法のことです。例えば「2009/10/22」といった日付を全て探したい、別の文字列に置換したい場合に用います。Windows用の無料テキストエディタで正規表現検索・置換ができるものとしてはサクラエディタがあります。
http://sakura-editor.sourceforge.net/
からダウンロードできます。インストーラをダウンロードして実行し、予めインストールしておいて下さい。
本書ではEMBOSSというソフトを利用します。Windows用のEMBOSSは
ftp://emboss.open-bio.org/pub/EMBOSS/windows/
でインストーラが配布されていますのでこれをダウンロードして起動し、指示通りにインストールすれば完了です。
次に、配列を表示するためのソフトとしてMEGAかJalviewをインストールして下さい。それぞれ下記のURLから入手できます。
http://www.megasoftware.net/
http://www.jalview.org/Web_Installers/install.htm
Jalviewはデフォルトでは起動時にデモが始まってしまい、それが非常に重いので、Toolsメニュー内のPreferences...を開き、Open fileのチェックボックスに入っているチェックを外して下さい。これでデモが起動しなくなります。
その他のソフトウェアは、
http://www.fifthdimension.jp/products/molphypack/
にインストーラを用意してあります。ダウンロードして実行し、案内に従ってインストールしておいて下さい。
分子系統樹推定で用いられるソフトウェアには、英語圏で制作されたものが多くあります。そのようなものはしばしば日本語の文字を含んだフォルダ名・ファイル名を正しく扱うことができません。しかし多くのOSでユーザー用のフォルダはユーザー名を含んでいます。そのため、ユーザー名に日本語(に限らず英数字以外の文字)を用いていると問題が起きる可能性があります。的確なエラーメッセージが表示されれば原因は分かるし対策も打てるのですが、エラーメッセージを見ても原因が分からないことも頻繁にありますので、もしユーザー名に英数字以外を用いていた場合、新たに英数字以外の文字をユーザー名に含まないアカウントを作成してそちらのアカウントでログオンするようにして下さい。特定のファイルやフォルダの最上位のフォルダからの位置を正確に記したものを絶対パスとかフルパスと言います。フルパスにスペースを含んでいると正常に動作しないソフトウェアもあるかもしれませんので注意して下さい。特に、Windows XPではデスクトップやマイドキュメントはフルパスにスペースが含まれていますので注意が必要です。
また、最近のOSには自動更新機能が搭載されていることがありますが、更新の際に強制的に再起動するものがあります。分子系統樹推定は非常に時間のかかる解析です。1ヶ月かかる解析の最中に強制再起動が働いて、また最初からやり直し、などということになっては大変です。例えば、Windowsでは更新を定期的に確認し、もし見つかれば自動的にインストールして、必要があれば強制再起動するのがデフォルト設定になっています。というわけで、強制的に再起動されたりすることの無いように設定を確認しておいて下さい。処理の重いスクリーンセイバーや常駐ソフトウェアも解析の邪魔になりますので、解析中は無効にしておくことをおすすめします。
0.2 Mac OS Xの場合
Mac OS Xは正統なUNIXの流れを汲むOSであり、ほとんどのUNIX環境用ソフトウェアがそのままで動作します。標準でターミナルがインストールされており、Java・Perlも入っています。しかし、Cコンパイラなどの重要なコマンドは標準ではインストールされていません。CコンパイラはXcode Toolsの一部としてAppleから提供されています。
https://developer.apple.com/downloads/index.action
から事前にダウンロードしてインストールしておく必要があります。開発者として登録した上で、OSのバージョンに合ったものをインストールする必要がありますのでご注意下さい。Snow Leopard以前のOSであれば、OSのインストール用DVDに同梱されているはずですので、それをインストールしていただければ結構です。なお、Lion以降では、オプションとして提供されているCommand Line Tools for Xcodeという名前のパッケージも必要です。こちらもインストールしておいて下さい。
後の操作の際に「正規表現検索・置換」に対応したテキストエディタがあると大変便利です。正規表現とは、「一定のルールに該当する文字列を検索する」ためのそのルールの記述方法のことです。例えば「2009/10/22」といった日付を全て探したい、別の文字列に置換したい場合に用います。Mac OS X用の無料テキストエディタで正規表現が利用可能なものとしては、CotEditorがおすすめです。CotEditorは下記から入手できます。
http://sourceforge.jp/projects/coteditor/
ダウンロードしてアプリケーション(/Applications)に入れておいて下さい。
Mac OS Xでは、多くの作業をターミナルで行いますが、ターミナル上でのフォルダの移動は面倒な作業です。以下のURLから「cdto」というソフトをインストールしておくことでその手間が軽減できます。
https://code.google.com/p/cdto/
配布ファイルを展開して、OSのバージョンに合った実行ファイルをアプリケーション(/Applications)にインストールして下さい。インストール後、Finder上でcdtoのアイコンをFinderのタイトルバー付近の適当な場所にドラッグアンドドロップして下さい。cdtoのボタンが登録されます。以降、そのボタンを押すことでFinderで開いているフォルダをカレントフォルダとするターミナルが起動できるようになります。
次に、配列を表示するためのソフトとしてMEGAかJalviewをインストールして下さい。それぞれ下記のURLから入手できます。
http://www.megasoftware.net/
http://www.jalview.org/Web_Installers/install.htm
Jalviewはデフォルトでは起動時にデモが始まってしまい、それが非常に重いので、Toolsメニュー内のPreferences...を開き、Open fileのチェックボックスに入っているチェックを外して下さい。これでデモが起動しなくなります。
以上のインストールが終わったら、以下のコマンドをターミナルで実行して下さい。必要なものが全てインストールされます。途中、何回か管理者パスワードを質問されますので、入力して下さい。
> mkdir -p ~/temporary↓ > cd ~/temporary↓ > curl -O http://www.fifthdimension.jp/products/molphypack/install_on_OSX.sh↓ > sh install_on_OSX.sh↓ > cd ..↓ > rm -rf temporary↓
もしも外部ネットワークへのアクセスにプロキシを設定する必要がある場合は、上記のコマンド実行の前に以下のコマンドを実行して環境変数を設定しておいて下さい。これにより、外部へはプロキシを経由してアクセスが行われるようになります。
> export http_proxy=http://server.address:portnumber↓ > export ftp_proxy=http://server.address:portnumber↓
なお、ユーザー名とパスワードを用いた認証が必要なプロキシでは、以下のようにして下さい。
> export http_proxy=http://username:password@server.address:portnumber↓ > export ftp_proxy=http://username:password@server.address:portnumber↓
0.3 Linuxの場合
Debianでは、sources.listを編集してcontribとnon-freeを有効にしておいて下さい。Ubuntuではuniverse・multiverseが相当しますが、最初から有効になっていますので編集の必要はありません。その上で、以下のコマンドをターミナルかコンソールで実行して下さい。必要なものが全てインストールされます。途中、何回か管理者パスワードを質問されますので、入力して下さい。
> mkdir -p ~/temporary↓ > cd ~/temporary↓ > wget -c http://www.fifthdimension.jp/products/molphypack/install_on_Debian.sh↓ > sh install_on_Debian.sh↓ > cd ..↓ > rm -rf temporary↓
もしも外部ネットワークへのアクセスにプロキシを設定する必要がある場合は、上記のコマンド実行の前に以下のコマンドを実行して環境変数を設定しておいて下さい。これにより、外部へはプロキシを経由してアクセスが行われるようになります。
> export http_proxy=http://server.address:portnumber↓ > export ftp_proxy=http://server.address:portnumber↓
なお、ユーザー名とパスワードを用いた認証が必要なプロキシでは、以下のようにして下さい。
> export http_proxy=http://username:password@server.address:portnumber↓ > export ftp_proxy=http://username:password@server.address:portnumber↓
本スクリプトではテキストエディタは追加されません。EmacsでもVimでもgEditでもKateでも、お好みものをお使い下さい。検索・置換をPerlのワンライナーで行なっていただいても構いません。
第1章 配列データセットの作成
1.1 配列データファイルの形式と相互変換
各種データベースやソフトウェアでは、様々なデータファイル形式が用いられており、利用時には相互に変換する必要がしばしば生じます。以下ではまず各種ファイル形式について簡単に解説した後、相互変換方法について述べます。
1.1.1 各ファイル形式の特徴
GenBank形式
Web上の配列データベースにおけるスタンダードなファイル形式です。配列データ以外に、その配列に関する様々な注釈(annotation)情報を加えることができます。それらの情報に基づいたデータの加工処理もソフトウェアを用いて簡単に行うことができるため大変便利です。人間にとってもプログラムにとっても可読性の高いファイル形式と言えるでしょう。最も単純な場合は以下のような形式です。
| LOCUS ABC1234 60 bp | DEFINITION TaxonA 18S small subunit ribosomal RNA gene, partial sequence. | ORIGIN | 1 AAAAAAAAAA AAAAAAAAAA AAAAAAAAAA AAAAAAAAAA AAAAAAAAAA AAAAAAAAAA | // | | LOCUS ABC1235 60 bp | DEFINITION TaxonB 18S small subunit ribosomal RNA gene, partial sequence. | ORIGIN | 1 AAAAAAAAAA AAAAAAAAAA AAAAAAAAAA AAAAAAAAAA AAAAAAAAAA AAAAAAAAAA | // | | LOCUS ABC1236 60 bp | DEFINITION TaxonC 18S small subunit ribosomal RNA gene, partial sequence. | ORIGIN | 1 AAAAAAAAAA AAAAAAAAAA AAAAAAAAAA AAAAAAAAAA AAAAAAAAAA AAAAAAAAAA | //
ほとんどの場合はもっと様々な情報を含んでいるので、これほどシンプルではありません。
FASTA形式
Web上の配列データベースは、この形式でのデータ出力にも対応していることが多いと思います。しかし、注釈(annotation)情報はありませんので、それらの情報を用いた加工を行いたい場合には不適です。また、塩基配列決定を行った場合には、波形の編集や複数の配列を結合(assemble)した後、このファイル形式に配列データを書き出すことが多いでしょう。ほとんどの多重配列エディタ(multiple sequence editor)においてもスタンダードなファイル形式であり、いずれのソフトにおいても入力の互換性は高いと言えます。実際の配列編集を行う際にはこのファイル形式で作業することが多いでしょう。ClustalW/Xでは配列データキャラクタとして?に対応していないため、もし?があるならNなどに置換しておく必要があります。以下に典型的なFASTA形式ファイルを示します。
| >TaxonA | AAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAA | >TaxonB | AAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAA | >TaxonC | AAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAA
Clustal形式
ClustalW/Xにおいて多重配列アライメント(multiple sequence alignment)を行った際に出力されるデフォルトファイル形式です。オプション設定により他の形式での出力も可能です。
| CLUSTAL 2.0.12 multiple sequence alignment | | | TaxonA AAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAA | TaxonB AAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAA | TaxonC AAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAA | ************************************************************
PHYLIP形式
系統解析ソフトウェアにおいて最も多く利用されているファイル形式の一つです。単純なファイル形式ですが方言が多くあり、解析ソフトウェアごとにマニュアルを良く読んで確認する必要があるのがやっかいです。配列名の文字数に制限があり、元々は10文字しか使えませんでした。しかし、これを拡張して配列名と配列の間をスペースで区切ることにして配列名の文字数制限を緩めたものも多く使われています。最大の問題は、元々の配列名文字数10文字の仕様では配列名と配列との間をスペースで区切る必要が無かったため、配列名が10文字ぴったりの場合に両者に互換性が無いことです。よって、この形式を用いる際には配列名を10文字以内にした上で必ず配列名と配列の間をスペースで区切るようにし、元々のPHYLIP形式の仕様に準拠したものとするのが安全です。閲覧・編集に適したinterleaved形式もあり、テキストエディタでの操作に適しています。PHYLIPでは配列内にはスペースが含まれていても問題ありませんが、ソフトウェアによっては配列は一続きの文字列であることを仮定しているものもあります。interleaved形式に対応していないソフトウェアもあります。また、1行空けてさらに同じ形式でデータを続けることで、ブートストラップリサンプリングしたりした多数のデータセットを1ファイルに格納することもできます。GenBank・Clustal・FASTAではそのようなことはできません。
non-interleavedとinterleavedの違いは実際のファイルの中身を見ていただくのが分かり易いでしょう。以下がnon-interleavedのPHYLIP形式配列ファイルです。
| 3 60 | TaxonA AAAAAAAAAA AAAAAAAAAA AAAAAAAAAA AAAAAAAAAA AAAAAAAAAA | AAAAAAAAAA | TaxonB AAAAAAAAAA AAAAAAAAAA AAAAAAAAAA AAAAAAAAAA AAAAAAAAAA | AAAAAAAAAA | TaxonC AAAAAAAAAA AAAAAAAAAA AAAAAAAAAA AAAAAAAAAA AAAAAAAAAA | AAAAAAAAAA
そして、これがinterleavedのPHYLIP形式ファイルです。
| 3 60 | TaxonA AAAAAAAAAA AAAAAAAAAA AAAAAAAAAA AAAAAAAAAA AAAAAAAAAA | TaxonB AAAAAAAAAA AAAAAAAAAA AAAAAAAAAA AAAAAAAAAA AAAAAAAAAA | TaxonC AAAAAAAAAA AAAAAAAAAA AAAAAAAAAA AAAAAAAAAA AAAAAAAAAA | | AAAAAAAAAA | AAAAAAAAAA | AAAAAAAAAA
どちらも50座位で折り返しているのですが、non-interleaved形式ではそれぞれの配列ごとに折り返しているのに対して、interleaved形式では全配列をセットで折り返しています。前述のようにinterleaved形式に対応していないソフトもありますが、non-interleavedなのに折り返しがあるファイルに対応していないソフトもありますので注意が必要です。
NEXUS形式
系統解析ソフトウェアにおいて最も多く利用されているもう一つのファイル形式です。様々な「ブロック」を記述することができ、対応しているソフトウェア用のコマンドを記述しておくことができます。その「ブロック」に非対応のソフトウェアではその中の内容は無視されますので通常問題は生じません。配列もDataブロックというブロック内に記述します。本形式にも閲覧・編集に適したinterleaved形式があり、テキストエディタでの操作に向いています。また、PHYLIP形式と同様、さらにDataブロックを作成することで、ブートストラップリサンプリングしたりした多数のデータセットを1ファイルに格納することもできます。GenBank・Clustal・FASTAではそのようなことはできません。
| #NEXUS | | Begin Data; | Dimensions NTax=3 NChar=60; | Format DataType=DNA Interleave Missing=? Gap=-; | Matrix | TaxonA AAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAA | TaxonB AAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAA | TaxonC AAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAA | | TaxonA AAAAAAAAAA | TaxonB AAAAAAAAAA | TaxonC AAAAAAAAAA | ; | End;
1.1.2 データ形式の相互変換
配列名には、基本的に英数字とアンダースコア以外は使わないようにした方が無難です。その他の特殊記号を用いていてうまくいかない場合には、一時的に特殊記号を他の文字列に置き換えておくとよいでしょう。しかし、そのような文字は解析ソフト側でも問題が発生しやすいのでできるだけ使用は避けましょう。
seqretによるデータ変換
seqretはEMBOSSに含まれている配列ファイル入出力コマンドです。ほとんどの形式に対応しており、配列形式の相互変換に便利です。対応形式の一覧は
http://emboss.sourceforge.net/docs/themes/SequenceFormats.html
にあります。入力ファイルをPHYLIP/NEXUS形式へ変換するには以下のようにコマンドを実行します。
> seqret 入力ファイル phylip::出力ファイル↓ > seqret 入力ファイル nexus::出力ファイル↓
入力ファイル形式がうまく認識されていないと思われる状況では、入力ファイル形式を以下のように指定することで改善することがあります。
> seqret fasta::入力ファイル phylip::出力ファイル↓
Phylogears2によるデータ変換
Phylogears2には、FASTA・NEXUS・PHYLIP・Treefinderの4形式の相互変換が可能なpgconvseqコマンドがあります。NEXUSとPHYLIPは多数のデータセットを1ファイルに格納できますが他の形式はそうではないので、多数のデータセットを格納しているNEXUSやPHYLIP形式をFASTAやTreefinder形式に変換する場合は、独自ルールで書き出します。FASTAの場合、データセット間に空行を設けます。Treefinderでは、% end of dataというコメントを間に挟みます。これらを正しく解釈できるソフト(Phylogears2の一部コマンドだけです)でしかこれらが多数のデータセットであることを認識できません。また、変換元のFASTA形式配列に空行があると、NEXUSやPHYLIP形式に出力した際に別データセットとされてしまうので注意が必要です。使い方は下記のようになります。
> pgconvseq --output=PHYLIP 入力ファイル 出力ファイル↓ > pgconvseq --output=NEXUS 入力ファイル 出力ファイル↓ > pgconvseq --output=TF 入力ファイル 出力ファイル↓
なお、PHYLIP形式では本来配列名は10文字以下でなくてはなりませんが、配列形式としてPHYLIPexを指定することで11文字以上の配列名も許容したファイルを作成することができます。PHYML・RAxML・PAMLでは、この形式で長いOTU名を使うことができます。
1.2 目的の配列を入手する
以下では配列データベースから目的の配列を探し出して得る方法について述べます。
1.2.1 分類群・遺伝子の名前から探す
配列が欲しい分類群が分かっているなら、分類群名データベースから辿ることで目的の配列を得ることができます。
まず、NCBI Taxonomyのサイトを開きます。URLは下記です。
http://www.ncbi.nlm.nih.gov/taxonomy/
このページで表示される検索ボックスから正式な分類群名で検索すると、データベース内で見つかった分類群のリストが出ますので、目的の分類群のリンクをクリックします。すると、高次分類群であれば所属する下位の分類群の階層化リストが出ます。最上位の分類群名をクリックすると、NCBIの他のデータベース内にある当該分類群のデータエントリ件数のリストが表示されています。高次分類群でなく種であればすぐにこの表示になります。件数にリンクが設定されていますのでクリックしてリンク先に跳ぶと、選択したデータベース内での当該分類群のデータエントリがずらっと出てきます。この状態で検索ボックスに遺伝子名などを追加すれば絞り込むことができます。NCBI Taxonomyを使わずとも、NucleotideやProteinのデータベースで分類群名で検索しても構いませんが、漏れや余計なものが入りやすいのでこちらの方法がおすすめです。
次に探したいデータの遺伝子名が分かっている場合です。この場合も分類群同様に遺伝子名データベースから辿ればよいでしょう。
まず、NCBI Geneのサイトを開きます。URLは下記になります。
http://www.ncbi.nlm.nih.gov/gene/
こちらの検索ボックスで目的の遺伝子名で検索します。ただ、それだけでは大量にヒットしてしまいますので、分類群名などを追加して絞り込むとよいでしょう。分類群名と同様、NucleotideやProteinのデータベースで遺伝子名で検索してもよいでしょう。他のデータベースへのリンクはあるものの分類群と違ってあまり役に立たないのでその方が手っ取り早いかもしれません。
NCBIのNucleotideやProteinのデータベースでは、それぞれのデータエントリにはそのデータ元の生物名、遺伝子名、配列長などの様々な情報が項目ごとに記載されています。ですから、それぞれの項目を指定してキーワード検索できれば余計なものが引っかかりにくくなったりして便利です。そのためには、以下のようなキーワードを書けばよいことになっています。
キーワード[項目指定語]
項目指定語の一覧は以下のURLで説明されています。
http://www.ncbi.nlm.nih.gov/books/NBK49540/
例えば、以下のようなキーワードを付加することで配列長が100~1,000のエントリのみに絞り込むことができます。
100:1000[Sequence Length]
これらの項目指定検索を組み合わせてやることで目的のエントリを見つけやすくなるでしょう。
目的のエントリが見つかったら、エントリ名をクリックすればいいですし、複数件ある場合は各エントリの頭にあるチェックボックスにチェックを入れてから検索結果リストの上にあるDisplayプルダウンメニューからGenBankを選択すれば、チェックを入れたエントリの生データ、即ちGenBank形式配列が表示されます。Showプルダウンメニューからは1ページに表示する件数、並び替えに使うもの(Sorted By)などを指定できます。Send toからはTextを選べばプレーンテキストで表示され、Fileならローカルファイルへ保存するダイアログが出るはずです。つまり、GenBank形式で表示している状態でSend toをFileにすれば、表示しているGenBank形式データをごっそり手元のマシンに保存できます。
1.2.2 配列から類似配列を探す
NCBI BLASTから配列データベース中の類似配列を探索することができます。URLは下記です。
http://www.ncbi.nlm.nih.gov/BLAST/
BLASTの基本的な使い方はライフサイエンス統合データベースプロジェクトが運営する統合TVにて動画で解説されていますのでそちらをご参照下さい。下記URLからアクセスできます。
http://togotv.dbcls.jp/
1.3 GenBank形式ファイルからの特定遺伝子配列の抽出
GenBank形式では、配列中のそれぞれの領域がどういうものかという注釈(annotation)が加えられています。この情報を利用すれば、長大な配列から特定の遺伝子領域のみを抽出することができます。
まず、GenBank形式のデータファイルをテキストエディタで開いてみて下さい。以下のような内容になっているはずです。
| LOCUS NC_001709 19517 bp DNA circular INV 06-MAY-2009 | DEFINITION Drosophila melanogaster mitochondrion, complete genome. | ACCESSION NC_001709 | VERSION NC_001709.1 GI:5835233 | DBLINK Project:164 | KEYWORDS . | SOURCE mitochondrion Drosophila melanogaster (fruit fly) | ORGANISM Drosophila melanogaster | Eukaryota; Metazoa; Arthropoda; Hexapoda; Insecta; Pterygota; | Neoptera; Endopterygota; Diptera; Brachycera; Muscomorpha; | Ephydroidea; Drosophilidae; Drosophila; Sophophora. | REFERENCE 1 (bases 1 to 408; 13319 to 19517) | AUTHORS Lewis,D.L., Farr,C.L. and Kaguni,L.S. | TITLE Drosophila melanogaster mitochondrial DNA: completion of the | nucleotide sequence and evolutionary comparisons | JOURNAL Insect Mol. Biol. 4 (4), 263-278 (1995) | PUBMED 8825764 | 略 | FEATURES Location/Qualifiers | source 1..19517 | /organism="Drosophila melanogaster" | /organelle="mitochondrion" | /mol_type="genomic DNA" | /db_xref="taxon:7227" | gene 1..65 | /gene="trnI" | /nomenclature="Official Symbol: mt:tRNA:I | Name: | mitochondrial isoleucine tRNA | Provided by: FBgn0013696" | /note="tRNA[Ile]" | /db_xref="FLYBASE:FBgn0013696" | /db_xref="GeneID:261011" | tRNA 1..65 | /gene="trnI" | /product="tRNA-Ile" | /db_xref="FLYBASE:FBgn0013696" | /db_xref="GeneID:261011" | 略 | gene 240..1263 | /gene="ND2" | /nomenclature="Official Symbol: mt:ND2 | Name: | mitochondrial NADH-ubiquinone oxidoreductase chain 2 | | Provided by: FBgn0013680" | /note="URF2" | /db_xref="FLYBASE:FBgn0013680" | /db_xref="GeneID:192474" | CDS 240..1263 | /gene="ND2" | /note="TAA stop codon is completed by the addition of 3' A | residues to the mRNA" | /codon_start=1 | /transl_except=(pos:1263,aa:TERM) | /transl_table=5 | /product="NADH dehydrogenase subunit 2" | /protein_id="NP_008277.1" | /db_xref="GI:5835234" | /db_xref="FLYBASE:FBgn0013680" | /db_xref="GeneID:192474" | /translation="MFNNSSKILFITIMIIGTLITVTSNSWLGAWMGLEINLLSFIPL | LSDNNNLMSTEASLKYFLTQVLASTVLLFSSILLMLKNNMNNEINESFTSMIIMSALL | LKSGAAPFHFWFPNMMEGLTWMNALMLMTWQKIAPLMLISYLNIKYLLLISVILSVII | GAIGGLNQTSLRKLMAFSSINHLGWMLSSLMISESIWLILFFFYSFLSFVLTFMFNIF | KLFHLNQLFSWFVNSKILKFTLFMNFLSLGGLPPFLGFLPKWLVIQQLTLCNQYFMLT | IMMMSTLITLFFYLRICYSAFMMNYFENNWIMKMNMNSINYNMYMIMTFFSIFGLFLI | SLFYFMF" | 略 | ORIGIN | 1 aatgaattgc ctgataaaaa ggattacctt gatagggtaa atcatgcagt tttctgcatt | 略 | //
これを見れば、FEATURESという項目にどこからどこまでが何という領域か、といった情報が書かれているのが分かります。ORIGINには実際の塩基配列があります。NCBI上のエントリをWebブラウザで見ている場合、FEATURESのCDSとかtRNAといった文字列にはリンクが設定されており、リンク先では該当領域だけが切り出して表示されます。領域を切り出したいエントリが少ない場合は、これを繰り返して切り出した情報を得ることもできますが、エントリ数が大きくなってくると手間がかかります。FEATURESの内容を任意のキーワードで検索して、該当する領域の配列をORIGINの内容から切り出してくれるコマンドextractfeatがEMBOSSに含まれていますので、これを使えば容易に大量のエントリから領域を切り出すことができます。
例えばtrnI領域を別ファイルに書き出すには、以下のようにターミナルやコマンドプロンプトでコマンドを実行します。
> extractfeat -type tRNA -tag gene -value trnI 入力ファイル 出力ファイル↓
このコマンドを実行すると、tRNA領域の中で遺伝子名にtrnIを含む領域が出力ファイルにFASTA形式で書き出されます。同様に、ND2領域を書き出すには以下のようにします。
> extractfeat -type CDS -tag gene -value ND2 入力ファイル 出力ファイル↓
データベースの注釈がきちんとなされていればこれでうまくいきますが、遺伝子名には微妙に表現が異なる記法が使われていることが頻繁にあります。そのような場合は、"ND2 | NAD2"などとスペースと|で区切って複数のキーワードを書き、ダブルクォートで囲ってやることでそれぞれのキーワードに一致する配列が出力されます。これは、複数の領域を一度に書き出したい場合にも使えます。 ただし、16S ribosomal RNAなどといった、上記のような区切り文字でないスペースを含んだキーワードは使用できません。 そのような場合は、事前に配列ファイルを正規表現を用いた検索・置換などを用いて処理しておきます。
また、書き出した領域を増幅できるプライマーを設計したい場合には、その領域の前後100bpほどまで含めて書き出したいことがあります。その場合には、以下のように-beforeオプションと-afterオプションを付加します。
> extractfeat -type CDS -tag gene -value ND2 -before 100 -after 100 入力ファイル 出力ファイル↓
1.4 多重配列整列
配列の準備ができたら、多重配列整列(multiple sequence alignment)によって各配列間で相同(homologous)な領域を検出して揃えてやる必要があります。これは、相同でない形質を比較しても系統樹の推定には役立たないためです。相同とは、「同じ祖先形質に由来する」という意味です。例えば、人間の眼と魚の眼は共通祖先が持っていた眼に由来すると考えられますが、イカやタコの眼はそうではありません。同様に、鳥の翼とコウモリの翼も相同ではありません。ただ、これらが相同でないというのは、我々が系統関係を知っているから分かるのであって、それが無ければそうとは分からないかもしれません。ですから、相同であるか否かと系統樹とは鶏と卵の関係に似ていると言えます。
配列の多重配列整列でも同じことが言えます。つまり、系統関係無しには正しい多重配列整列ができないのです。そこで、多重配列整列と系統樹推定を同時にやってしまおうという動きもあります(例えば Fleissner et al., 2005; Lunter et al., 2005; Redelings and Suchard, 2005, など)が、膨大な計算を要し、今のところ現実的ではありません。そこで、我々はそこそこ悪くないだろうと思われる「仮の系統樹」を作成し、それに基づいて多重配列整列を行い、系統関係に依存していると考えられる信頼性の低い領域は除去して系統樹推定に用いることにしています。
多重配列整列に最もよく用いられているのが、ClustalW2/X2 (Larkin et al., 2007)ですが、最近はMUSCLE (Edgar, 2004)やMAFFT (Katoh et al., 2005)という高速性や正確性で上回るプログラムが登場し、徐々にこれらへの移行が起きつつあります。ここではMAFFTを用いた多重配列整列の方法を説明します。
MAFFTはコマンドラインから実行するプログラムです。使用するには、コマンドプロンプトやターミナルで以下のようにします。入力ファイル・出力ファイル共にFASTA形式です。
> mafft --auto 入力ファイル > 出力ファイル↓
--autoオプションでは、MAFFTが備えているいくつかのアルゴリズムからデータサイズなどに応じて最適なものを自動的に選択してくれます。終了の際のメッセージにどのアルゴリズム(L-INS-i・E-INS-i・G-INS-i・FFT-NS-i・FFT-NS-2など)を用いたのかが表示されますので、論文にする際にはどれが使われたのかできるだけ書いた方が良いでしょう。
1.4.1 タンパクコード塩基配列の多重配列整列
タンパクコード塩基配列を塩基配列のままで整列すると、翻訳後のアミノ酸の変異を考慮していないため、容易にフレームシフトを起こすギャップが挿入されてしまいます。しかし、現実にはそんな整列結果が妥当であることはほとんどありません。また、遺伝暗号やアミノ酸の物理化学的性質上、起こりやすい・起こりにくい変異はかなり情報が蓄積されていますが、塩基配列の整列ではそのようなことも考慮されません。そこで、いったんアミノ酸配列に翻訳して整列してから、それを逆翻訳(正確には整列済アミノ酸配列を参照しながら塩基配列を整列)してやることで、多くの場合ただ単純に整列するよりも良い結果が得られます。ここでは多重配列整列にMAFFTを、逆翻訳にEMBOSSに含まれているtranalignを用いる方法を説明します。
まず、翻訳するには各配列でコドン位置が揃っている必要があるため、塩基配列のままで整列をします。
> mafft --auto 入力ファイル > 出力ファイル↓
整列したファイルをJalviewやMEGAなどで表示して見てやると、大抵の場合第3コドン位置では同義置換ばかりで他のコドン位置よりも変異が激しいためすぐに分かります。変異の多い座位が3座位ごとにあるわけです。そこで、第1コドン位置が1座位目になるように編集して保存します。もし途中から非コード配列になるようであればその領域も削除しておきます。翻訳してから削除しても構いません。もしもコドン位置が分からなかったり、翻訳の向きが分からなかったら、以下のようにEMBOSSのsixpackコマンドを使います。
> sixpack 入力ファイル↓
コマンドを実行すると保存先のファイルを聞かれるので適当に名前を付けるかデフォルトのままで保存します。ここで、遺伝暗号がstandardではない場合は、-tableオプションでそれを指示してやる必要があります。例えば昆虫のミトゲノム配列であればinvertebrate mitochondrialなので以下のようにコマンドを実行します。
> sixpack -table 5 入力ファイル↓
-tableオプションに指定する番号と遺伝暗号との対応は以下のようになっています。
-
0.
Standard (default)
-
1.
Standard with alternative initiation codons
-
2.
Vertebrate Mitochondrial
-
3.
Yeast Mitochondrial
-
4.
Mold, Protozoan, Coelenterate Mitochondrial and Mycoplasma/Spiroplasma
-
5.
Invertebrate Mitochondrial
-
6.
Ciliate Macronuclear and Dasycladacean
-
9.
Echinoderm Mitochondrial
-
10.
Euplotid Nuclear
-
11.
Bacterial
-
12.
Alternative Yeast Nuclear
-
13.
Ascidian Mitochondrial
-
14.
Flatworm Mitochondrial
-
15.
Blepharisma Macronuclear
-
16.
Chlorophycean Mitochondrial
-
21.
Trematode Mitochondrial
-
22.
Scenedesmus obliquus
-
23.
Thraustochytrium Mitochondrial
sixpackコマンドで出力されるファイルのうち、FASTA形式配列の方を開くと、入力ファイルの1つ目の配列で順方向3フレーム、逆方向3フレームの全6フレームでの翻訳がなされた結果得られたopen reading frame (ORF)の配列が保存されています。ORFとは、開始コドンから終止コドンまでの配列です(ここでは実際には終止コドンで区切っただけの配列となっています)。これが最も長くなるのが正しい翻訳結果と考えられます。sixpackコマンドで出力されるもう一つのファイルには、入力ファイルの1つ目の配列で6フレーム翻訳を行った結果がテキストエディタで見やすく出力されていますのでこちらでも確認できます。末尾に6フレームそれぞれでできるORF数がありますので、これが少ない方が正しい可能性が高いでしょう。もし読み枠が逆方向だったら、revseqコマンドで必要に応じて逆相補配列に変換することができます。
ファイルに含まれる塩基配列が複数あり、その解読方向が統一されていない場合には、Phylogearsのpgstanstrandコマンドによって先頭の配列と同じ方向に揃ったファイルを得ることができます。以下のように使用して下さい。
> pgstanstrand 入力ファイル 出力ファイル↓
ただし、このコマンドはFASTA形式にしか対応していませんのでご注意下さい。
正しくコドン位置を揃えることができたら、そのファイルから以下のようにEMBOSSのdegapseqコマンドでギャップを除去してやります。
> degapseq 入力ファイル 出力ファイル↓
ギャップを除去したら、以下のようにEMBOSSのtranseqコマンドを用いてアミノ酸配列に翻訳してやります。ここでもstandard以外の遺伝暗号の場合は-tableオプションで遺伝暗号を指定してやって下さい。
> transeq 入力ファイル 出力ファイル↓
翻訳したアミノ酸配列ファイルを念のためテキストエディタや多重配列エディタで開いて確認したら、以下のようにMAFFTで整列します。
> mafft --auto 入力ファイル > 出力ファイル↓
そして、最後にEMBOSSのtranalignコマンドでアミノ酸配列から元の塩基配列へ逆翻訳してやります。ここでもstandard以外の遺伝暗号の場合は-tableオプションで遺伝暗号を指示する必要があります。
> tranalign 未整列塩基配列ファイル 整列済アミノ酸配列ファイル 出力ファイル↓
なお、ここで述べたようにアミノ酸配列の整列に合わせて塩基配列を整列することが常に良いとは限りません。複数回のフレームシフトが起きている場合には塩基のまま整列した方が良いこともあります。そのため、塩基のまま整列した結果と必ず比較して確認するようにして下さい。また、読み間違いに起因すると思われるindelが存在する場合には、一時的に仮の塩基を挿入して整列してから、最終的なファイルでその塩基を?や-にする必要があります。
1.5 分子系統樹推定に不適な領域の除去
大抵の整列した配列データには、そのままでは分子系統樹推定に不適な部位を含んでいます。そのため、そのような部位を除去してから系統樹推定に用いる必要があります。ここでは系統樹推定に適している・適していないとはどういうデータかを説明した上で、その他の注意点を述べます。
1.5.1 オルソロガスとパラロガス
整列によって得られた相同(homologous)な配列データセットでも、系統樹推定に使えるとは限りません。例えば図1.1のようにY locusとZ locusが遺伝子重複によって生じた場合を想定すると、Taxon AのY locusとTaxon BのY locus、Taxon CのZ locusは相同ではありますが、正しい系統関係(Taxon BとTaxon Cが単系統でTaxon Aはその外側)を推定することはできません。このような関係をパラロガス(paralogous)と言います。それに対してそれぞれのY locusどうしやZ locusどうしの関係はオルソロガス(orthologous)と呼ばれています。系統樹推定には、オルソロガスなデータセットを用いなければなりません。ですから、遺伝子重複が起きたことが分かっている領域はできるだけ系統樹推定には使わない方が無難です。ただし、重複した遺伝子の全配列を全てのOTUで揃えれば、正しい系統樹を推定可能です。ある程度なら配列が欠けていても何とかなる場合もあるでしょう。

問題は遺伝子重複が起きたかどうかをどうやって知るかですが、これは近縁種で全ゲノムデータが得られていれば、ゲノム内BLASTで一致度の高い複数の領域が見つからないことを確認すればよいでしょう。BLASTの方が確実だと思いますが、Ensembl genome browserに登録されていれば、こちらでも確認することができます。Ensemblのサイトは以下のURLからアクセスして下さい。
http://www.ensembl.org/
近縁種のゲノムで重複が見つからないからといってオルソロガスとは言い切れませんが、これ以上は確認のしようがないので致し方ないでしょう。より多くの領域を用いて系統樹推定することで信頼性を担保する以外に無いと思います。
その他、incomplete lineage sortingや遺伝子水平伝播によっても、遺伝子の系統樹が種の系統樹と異なってしまうことがあります。例えばαとβが近縁(単系統)でγはその外に位置する系統関係にある3種α・β・γがいるとき、共通祖先時代に既に分化していた対立遺伝子A・aがあるとします。αとγではAが偶然固定し、βではaが偶然固定したとすると、Aとaの配列に基づく系統樹ではαとγが近縁(単系統)になってしまいます。これがincomplete lineage sortingです。また、incomplete lineage sortingによって生じた類似性をhemiplasyと言います(Avise and Robinson, 2008)。全くの別系統で収斂や平行進化で生じる類似性をhomoplasyと言いますが、それに合わせて最近提案された用語です。遺伝子水平伝播はその名の通り、何らかの作用によりある生物の遺伝子が、全く別系統の生物のゲノム内に取り込まれてしまうことです。この場合も遺伝子の系統樹は種の系統樹と一致しなくなってしまいます。
1.5.2 仮定を満たしていないデータ
分子系統樹推定は、様々な仮定を置いて適当にでっち上げた基準で系統樹を評価し、最も良いものを選ぶというものです。ですから、基準そのものの妥当性はさておき、その基準できちんと評価をするにはデータが仮定を満たしている必要があります。この仮定は、最節約法よりも最尤法やベイズ法などのモデルベースの方法の方がより多くなっています。
まず、全ての方法で共通な仮定として、「1座位の塩基・アミノ酸から1座位の塩基・アミノ酸への変異しか含まない」というものがあります(コドン置換モデルの場合は「1つのコドンから1つのコドン」)。分子進化モデルは1座位のアミノ酸から複数座位のアミノ酸への変異など想定していませんし、この仮定を満たしていないと最節約法でも変化の回数を過大評価してしまいます。具体的には、開始・終止コドンから別のコドンへの変異とその逆(1コドンから複数コドンへの変異とその逆)、イントロン両端のスプライセオソーム認識配列から別の配列への変異とその逆(非コード配列=0コドンから複数コドンへの変異とその逆)、フレームシフト・逆位(複数座位から複数座位への変異)、挿入・欠失(無から有とその逆)がそれに当たります。ただし、挿入・欠失は整列が信頼できるならギャップをただのmissing dataとして取り扱うことで対処できます(ほとんどのソフトウェアがそういう実装になっています)。最節約法では、ギャップを第5 (アミノ酸では第21)の形質状態として取り扱うことも可能ですが、一度の挿入・欠失で生じた連続したギャップが、複数回の挿入・欠失で生じたと解釈されてしまうため、おすすめできません。
次に、モデルベースの方法が仮定しているものとして「系統樹上で分子進化パターンが共通である」というものがあります。現状の分子系統樹推定法では系統樹全体で共通の分子進化モデルを当てはめているからです。ただ、そのような仮定をせずに系統樹上で分子進化モデルを変化させることが可能な推定方法もあるにはある(例えば Boussau and Gouy, 2006; Blanquart and Lartillot, 2006, 2008, など)のですが、計算量が膨大だったりするため現状ではほとんど使われていません。遺伝暗号やコドン使用頻度がOTU間で共通でないタンパクコード塩基配列はこれらの仮定を満たしていない可能性が極めて高いのでそのようなデータからモデルベースの方法で系統樹推定を行うのは避けた方が良いでしょう。また、塩基・アミノ酸頻度がOTU間で共通でない塩基・アミノ酸配列も同様です。塩基・アミノ酸頻度がOTU間で共通でない塩基・アミノ酸配列は、RY coding (Woese et al., 1991)やDayhoff coding (Hrdy et al., 2004)を用いて情報を多少捨ててでも無理矢理塩基・アミノ酸頻度を共通にしてしまうか、不均質モデル(Blanquart and Lartillot, 2006, 2008)を当てはめれば解析は可能です。
最後に、既に述べたことともやや重複しますが、同じ分子進化モデルを当てはめた座位間では同じ分子進化パターンに従っていなくてはなりません。ですから、座位ごとに分子進化パターンが異なると予想される場合(異なる遺伝子座など)には、異なる分子進化モデルを各座位に当てはめるべきです。しかし、異なる分子進化モデルを当てはめれば推定しなくてはならないパラメータが増加してしまいます。開始・終止コドンや、複数の遺伝子に共有されている座位の配列は他とは明らかに異なる選択圧にさらされているはずですから、当然分子進化パターンは異なると予想されます。とは言え、わざわざパラメータ数を増やしてまで異なるモデルを当てはめるほどの情報量は持っていないでしょうから、そのような座位は捨てた方が無難でしょう。
1.5.3 整列の信頼できない座位
偽遺伝子や遺伝子間領域、イントロン、rRNA/tRNAのloop領域などの欠失や挿入の多い配列では、整列の信頼性が低くなってしまいます。誤って整列された座位は、系統樹推定の際のノイズとなってしまうため、除去した方がよいと言われています(Talavera and Castresana, 2007)。これまでのところ、そのような処理が研究者の経験と勘でなされることが多かったのですが、近年になって自動的に行ってくれるソフトウェアが登場してきました。それがGblocks (Castresana, 2000)・trimAl (Capella-Gutiérrez et al., 2009)・Aliscore (Misof and Misof, 2009)・BMGE (Criscuolo and Gribaldo, 2010)です。ここではtrimAlを用いて整列の信頼できない座位をトリミングする手順を説明します。
trimAlが対応している入力ファイル形式はPHYLIP・FASTA・NEXUSなどです。trimAlでは、様々なパラメータをユーザーが設定することもできますが、ギャップをそれなりに残す設定とギャップを残さない設定、さらにその2つからデータに応じて自動的に選択させることもできます。それぞれの設定によるトリミングは以下のようにして行います。
> trimal -gappyout -in 入力ファイル -out 出力ファイル↓ > trimal -strict -in 入力ファイル -out 出力ファイル↓ > trimal -automated1 -in 入力ファイル -out 出力ファイル↓
ただし、タンパクコード塩基配列では読み枠がずれないように、コドン単位でのトリミングをする必要があります。trimAlはそこまで考えて処理をしてくれませんが、Phylogears2のpgtrimalコマンドを用いることでそれが可能です。pgtrimalは内部でtrimAlを呼び出して除去しない座位を得た上で、読み枠がずれないように除去する範囲を拡大します。入力ファイルはNEXUS形式でなくてはなりません。以下のようにして用います。
> pgtrimal --frame=1 --method=gappyout 入力ファイル 出力ファイル↓ > pgtrimal --frame=1 --method=strict 入力ファイル 出力ファイル↓ > pgtrimal --frame=1 --method=automated1 入力ファイル 出力ファイル↓
pgtrimalは--frameオプションがあると入力ファイルをタンパクコード塩基配列として扱います。--frame=1は配列の1塩基目が第1コドン位置であるという意味です。--frame=2であれば2塩基目が、--frame=3であれば3塩基目が第1コドン位置であるということになります。
1.5.4 その他の注意点
塩基配列データは、昔はRI、現在は蛍光や電位の変化を検出することで得ているはずです。そのようなデータは、検出されたシグナル強度の波形から読み取られているでしょう。しかし、しばしば波形が重なっていてどの塩基か特定できないことがあります。特に核ゲノムの配列をクローニングせずに直接読んでいる場合にヘテロな個体でよくあることだと思います。このような場合、解析ソフトは表1.1のような縮重コード表記を考慮してくれますので、何でもすぐにNにせずにRやYも積極的に用いた方が良いと思います。ただし、そのような不確実なデータを使わないのが最も安全ではあります。また、ギャップやギャップかどうかもよく分からないmissing dataはそれぞれ-・?として区別できるようにしておいた方が良いでしょう。
| 文字 | 意味 |
|---|---|
| M | A or C (amino) |
| R | A or G (purine) |
| W | A or T |
| S | C or G |
| Y | C or T (pyrimidine) |
| K | G or T (keto) |
| V | A or C or G |
| H | A or C or T |
| D | A or G or T |
| B | C or G or T |
| N | A or C or G or T |
タンパクコード塩基配列の編集の際には、必ず読み枠と翻訳後のアミノ酸配列が変化しないように注意して下さい。読み枠がずれると、コドン位置ごとの異なるモデルの当てはめがうまくいきません。第2・3コドン位置と次のコドンの第1コドン位置を削除すると、読み枠はずれませんが、後になってアミノ酸配列に変換する必要が生じたときやコドン置換モデルを当てはめようとした場合にうまくいかなくなってしまいますし、ケアレスミスの元なのでこれも避けるべきです。
配列の編集では、削除した座位がすぐに分かるように記録を残しておくとやり直したり削除した座位を確認したりする際に役立ち、ミスを防いだりミスに気付きやすくなります。実際に解析に用いる配列ファイルとは別に、削除した座位を[]などで囲んだファイルを別に保存しておくとよいでしょう。グラフィカルインターフェイスを持った多重整列エディタは便利ですが、そのような記録を残す機能を持っていないものがほとんどでしょうから、個人的には画面内に収まるように配列を折り返したinterleaved形式で保存したファイルをテキストエディタで編集するのが最も良いと思います。多重整列エディタで編集したい場合は、少なくとも何も削除していないファイルも保存しておき、削除後のファイルの配列と比較すればすぐに削除した部分が分かるようにしておくべきでしょう。
また、この先用いる解析ソフトでは、配列名には半角英数字とアンダースコア(_)しか使わない方が無難です。他の文字列を用いていたら、必ず別の配列が同一の名前にならないように注意しながら削除しておきます。形質が最初の配列と同じであることをピリオド(.)で表す方法がありますが、これも使わない方が安全です。対応したソフトで別形式で書き出すなどして無くしておきましょう。ファイル名にも注意が必要です。やはり半角英数字とアンダースコアしか使わないようにした方が良いでしょう。
1.6 配列が完全一致するOTUの除去
系統解析では配列が完全に一致する複数のOTU (系統樹末端の生物およびその配列)を含んでいると、そのOTUが他のOTUより大きく評価されることになり、推定結果に悪影響を及ぼしてしまいます。これをnode density artifactと言います(Webster et al., 2003; Venditti et al., 2006)。そのため、完全一致する配列はただ1つを残して他は除いておく必要があります。Phylogears2のpgelimdupseqコマンドを用いることで簡単に処理できます。以下のように用います。
> pgelimdupseq --type=DNA 入力ファイル 出力ファイル↓
アミノ酸配列では--type=DNAの代わりに--type=AAを指定して下さい。これによって完全一致する配列はただ1つを残して取り除かれます。残される配列の配列名(OTU名)は、除去された配列の名前を2連続のアンダースコア「__」で連結したものとなります。FASTA・NEXUS・PHYLIP・extended PHYLIP・Treefinder形式の入力ファイルに対応しています。ただし、PHYLIP形式は配列名が10文字までしか使えませんので特殊な処理を行っています。
ここで、縮重コード文字の取り扱いが問題になってきます。塩基配列では「AまたはG」という意味で「R」を用います。「AまたはCまたはGまたはT」の場合は「N」となります。この縮重コード文字がデータに含まれているときに、縮重コード文字をそのままにして全形質が一致しているものだけを完全一致配列とするのか、縮重コード文字を本来の意味通り「AまたはG」などと解釈して完全一致配列を探すのか、がまず問題となります。筆者の個人的な意見では後者が妥当であろうと思います。
後者を採用した場合、残す配列では形質を「A」とするのか「R」とするのかがさらなる問題となります。例えば「AAA」と「ARA」という配列があった場合、これらは完全一致となりますが、どちらを残すべきかということです。「R」が塩基配列決定の信頼性が低いために「R」とされているなら、残すのは「AAA」でよいでしょう。「R」となっている原因がノイズであり、ノイズを捨てることは何ら問題ではないからです。しかし、核DNAを多数クローンで配列決定を行いコンセンサス配列をデータとしている、または核DNAをクローニングせずに直接配列決定して「A」と「G」の両方のシグナルが検出されたために「R」としているのであれば、「ARA」にすべきかもしれません。「R」はノイズによるのではなく意味があるのですから。ただし、「R」には意味があるというのであれば、(あまり好ましくありませんが)「AAA」と「ARA」はやはり両方残すべきということになるかもしれません。pgelimdupseqは、標準では「AAA」を残します。「ARA」を残したい場合は--prefer=degenerateというオプションを入力ファイル名の前に付けて実行して下さい。両方を残したい場合は--prefer=bothとします。筆者はpgelimdupseqの標準設定を強く推奨します。なお、pgelimdupseqはギャップを意味する「-」を「?」 (missing data,「-またはN」の意)として取り扱います。ギャップを意味のある形質として取り扱うには、--gap=anotherをオプションとして指定します。
なお、完全一致しない場合でも、ごく近縁な配列が一部の系統でのみやたらと密にサンプリングされている場合にも、同一配列が複数登録されているのと同様の効果を発揮してしまいます。したがって、全種をサンプリングするのが必ずしも良くないこともあり得ます。理想的なのは、系統樹上の「分岐点密度」が全体に均一である、あるいは全ての枝の長さが均一であることです。実際にはほとんどそんなことはないでしょうが、できればそのようにタクソンサンプリングがなされることが望ましいでしょう。
1.7 塩基・アミノ酸組成の均一性の検定とデータ改変による均一化
ほとんどの分子進化モデルでは、塩基組成やアミノ酸組成はOTU間で均一であることが仮定されています。ですから、解析対象のデータがその仮定を満たしているかどうかは解析結果に大きな影響を及ぼします。塩基組成やアミノ酸組成がOTU間で均一でない場合、本当は単系統ではないOTU群の単系統性が非常に強く支持されてしまうことがしばしばあります。そのような、仮定を満たしていないデータに基づいてあり得ない単系統性を見いだしている論文が公表されることが未だに後を絶ちません。データ配列において塩基組成・アミノ酸組成の均一性が棄却されないことを確認しておけば、そのような論文を公表せずに済むはずです。Kakusan4・Aminosanもモデル選択前にこの検定を行いますが、Phylogears2に含まれているpgtestcompositionを用いることで、検定だけを行うことができます。
組成の均一性を検証するにはいくつかの方法がありますが、pgtestcompositionではχ二乗統計量を用いた独立性の検定を利用します。「組成は均一である」が帰無仮説です。これはPAUP*(Swofford, 2003)のBaseFreqsコマンドに実装されているのと同じ方法です。ただしPAUP*では塩基配列にしか適用できませんがpgtestcompositionではアミノ酸配列にも適用できます。また、PAUP*は「R」なら「A」と「G」がそれぞれ0.5回出現などとしてカウントすることで、縮重コード文字を検定統計量の算出に利用しますが、pgtestcompositionは縮重コード文字を一切用いません。この検定法よりも良いとされているBowkerの検定というものもあります(Ababneh et al., 2006)が、その方法ではある条件下ではp値を算出できず、その条件を満たすデータがしばしばあるため今のところは独立性の検定を利用しています。pgtestcompositionでこの検定を行うには、以下のようにコマンドを実行します。
> pgtestcomposition --type=DNA 入力ファイル 出力ファイル↓
アミノ酸配列では--type=DNAを--type=AAに置き換えて下さい。対応している入力ファイル形式はFASTA・NEXUS・PHYLIP・extended PHYLIP・Treefinderです。出力ファイルには以下のような情報が出力されます。
| Type of Nucleotides: 4 | Number of Taxa: 8 | Degree of Freedom: 21 | Total Count: 15994 | Chi-square Statistic: 3.62583123080048 | p-value: 0.99999 | | A C G T rtotal | OTU名 781 163 234 821 1999 | 770.65 171.73 236.22 820.40 | | 略 | | ctotal 6166 1374 1890 6564 15994
もしも均一性が棄却されてしまった場合、データを改変することで無理矢理均一化してしまうか、組成の不均一性を許容するモデル(Blanquart and Lartillot, 2006, 2008)を適用した解析を行う必要があります。また、データによっては正確なp値が算出できないものがあります(Cochran, 1954)。そのようなデータではファイルの末尾にその旨が出力されます。また、この方法では配列が長い場合には過剰に均一性が棄却されやすくなってしまいますので、そのようなデータの取り扱いには注意が必要です。
タンパクコード塩基配列データの第3コドン位置や、多遺伝子座配列データのある1遺伝子座といった、データの一部分のみに範囲を絞って検定を行うこともできます。例えば以下のコマンドでは、入力ファイルの1~100塩基目の範囲だけで検定を行います。
> pgtestcomposition --type=DNA "1-100" 入力ファイル 出力ファイル↓
第3コドン位置のみを対象としたい場合には以下のようにします。
> pgtestcomposition --type=DNA "3-.\3" 入力ファイル 出力ファイル↓
ここで、3-.\3は、3塩基目から末尾までの範囲において3塩基ごとに(2塩基間隔で)対象となる座位を選択するという意味です。なお、LinuxやMac OS Xなどのターミナル上では、3-.\3をダブルクオートまたはシングルクオートで囲むか、3-.\\3とタイプする必要があります。これは、ターミナル上ではクオートされていない\は特殊な意味のある文字だからです。ただし\\と記述することでコマンドに\を含む文字列を渡すことができます。同様に、?や*もターミナルでは特殊な意味があるので、コマンドにそのまま渡すにはクオートしておくか\を直前に付ける(これを「エスケープする」と言う)必要があります。
組成の均一性が棄却されてしまった場合、データを改変することで無理矢理均一化することができます。代表的な方法にRYコーディング(Woese et al., 1991)があります。RYコーディングは、塩基配列を対象としたデータ変換の方法です。塩基配列において組成が不均一なのは、ATとGCの比率がOTUによって異なるためであることがよくあります。このようなデータであっても、AGとCTの比率ははOTU間で均一になっている場合があります。これを利用すれば、形質状態を表す文字を「AまたはG」(つまり「R」)を表す文字と、「TまたはC」(つまり「Y」)を表す文字の2つだけにしてしまうことで、組成を均一化できます。この方法ではAG間、およびTC間の変異の情報は捨ててしまうことになりますが、従来の系統樹推定法をそのまま利用できます。Phylogears2では、pgrecodeseqコマンドを用いることでこの処理が容易に可能です。以下のコマンドを実行することで、RYコーディングを適用した配列を得ることができます。
> pgrecodeseq --type=DNA "CG-TA" 入力ファイル 出力ファイル↓
このコマンドを実行すると、「C」は「T」へ、「G」は「A」へそれぞれ置換された配列が出力されます。このため、配列は「A」と「T」の2文字だけになります(ただし縮重コード文字や「-」と「?」は除く)。RYの2文字になるわけではありませんが、効果は同じです。CG-TAの代わりにC-Tを指定すれば、「C」が「T」へ置換されるだけなので、AGYコーディングということになります。対応している入力ファイル形式はFASTA・NEXUS・PHYLIP・extended PHYLIP・Treefinderです。アミノ酸配列に対して用いられるDayhoffコーディング(Hrdy et al., 2004)というものもありますが、これは以下のように実行することで適用できます。
> pgrecodeseq --type=AA "STGPNEQKHVILYW-AAAADDDRRMMMFF" 入力ファイル 出力ファイル↓
これにより、出力される配列は「ADRMFC」の6文字だけになります。変換後のデータはRAxML (Stamatakis, 2006)やTreefinder (Jobb et al., 2004)、MrBayes (Ronquist and Huelsenbeck, 2003)で一般時間反転可能(GTR)モデルを適用して解析することができます。絶対にWAG (Whelan and Goldman, 2001)やJTT (Jones et al., 1992)などの経験的置換モデルやそれらの+Fモデルを適用してはいけません。従って、Dayhoffコーディングを行ったデータではモデル選択は必要ありません。pgrecodeseqは縮重コード文字も適切に処理するように作成してありますので、縮重コード文字の含まれている配列にもお使いいただけます。ただし、置換前の文字列と置換後の文字列には縮重コード文字を用いることはできませんのでご注意下さい。これはプログラム作成上の都合と、pgtestcompositionが縮重コード文字を統計量の計算に用いないためです。
タンパクコード塩基配列データの第3コドン位置や、多遺伝子座配列データのある1遺伝子座といった、データの一部分のみにこの処理を適用することもできます。範囲の指定方法はpgtestcompositionと同様です。データの一部分にのみ均一化の処理を行った場合、処理した部分と処理していない部分は異なるパーティションとし、比例または分離モデルを適用する必要があります。そうしないと、塩基組成や置換速度のパラメータが正しく推定できなくなるためです。また、データの変換ができたら、pgtestcompositionを用いて組成がOTU間で均一になっていることを確認してから実際の解析に用いるようにご注意下さい。
なお、上記のようなRYコード化したデータをRAxMLで解析しようとすると、「C」や「G」が存在しない塩基配列データになってしまうので、解析がうまくいかなくなります。そのような場合は「AT」を「01」に置き換えて下さい。
> pgrecodeseq --type=ANY "ATMWSKVHDBN-01?????????" 入力ファイル 出力ファイル↓
Dayhoffコーディングの場合は、元のアミノ酸配列を以下のようにして数字へ変換します。
> pgrecodeseq --type=ANY "ARNDCQEGHILKMFPSTWYVX-01223220144145000554?" 入力ファイル 出力ファイル↓
ただし、これらの場合はRAxMLではMULTIGAMMAモデルを当てはめる必要があります(01データの場合はBINGAMMAでも構いません)。-mオプションやパーティションの設定ファイルでMULTIGAMMAを指定して下さい。-KオプションはGTRにする必要がありますが、デフォルトでそうなっているはずです。万一MKになっていたらGTRに変更して下さい。GTRでは、ある座位の0と別の座位の0が同じものを意味することになります。MKでは、ある座位の0と別の座位の0が同じものを意味しないことになります。この設定は全てのパーティションに適用され、特定のパーティションではGTR、別のパーティションではMKといった設定にすることはできません。
第2章 分子進化モデルの基礎
分子進化モデルは塩基配列データに当てはめられる塩基置換モデル(nucleotide substitution model)と、アミノ酸配列データに当てはめられるアミノ酸置換モデル(amino acid substitution model)に大別されます。タンパクコード塩基配列データにおいて同義置換(synonymous substitution)と非同義置換(nonsynonymous substitution)を区別するコドン置換モデル(codon substitution model)というものもあります。コドン置換モデルはパラメータが多く計算が大変なのと対応しているソフトウェアが少ないため今のところあまり使われていませんが、よりリアルな確率過程を表しているため、将来的にはタンパクコード領域ではコドン置換モデルが多用されるようになっていく可能性は高いでしょう。しかし、ここでは塩基置換モデルとアミノ酸置換モデルに絞って説明を進めていきます。
2.1 塩基置換モデル
2.1.1 塩基置換速度行列
塩基置換速度行列(nucleotide substitution rate matrix)は、座位(site)内における、形質状態(character state)間の移行速度の不均質性(heterogeneity)を表現するものです。表2.1のように表すことができます。
| From\To | A | C | G | T |
|---|---|---|---|---|
| A | - | RateAC FreqC | RateAG FreqG | RateAT FreqT |
| C | RateAC FreqA | - | RateCG FreqG | RateCT FreqT |
| G | RateAG FreqA | RateCG FreqC | - | RateGT FreqT |
| T | RateAT FreqA | RateCT FreqC | RateGT FreqG | - |
ここで、RateXY FreqXは塩基Yから塩基Xへの移行速度で、FreqXは塩基Xの頻度です。ただし、RateXY = RateYXとします(これを「時間反転可能」(time-reversible)と言います)。
RateAC = RateAG = RateAT = RateCG = RateCT = RateGTであり、かつFreqA = FreqC = FreqG = FreqTのとき、最も単純なJC69モデル(Jukes and Cantor, 1969)となります。RateAG = RateCT ≠ RateAC = RateAT = RateCG = RateGTであり、かつFreqA = FreqC = FreqG = FreqTなモデルはK80/K2Pモデル(Kimura, 1980)です。RateAC = RateAG = RateAT = RateCG = RateCT = RateGTであり、かつFreqA ≠ FreqC ≠ FreqG ≠ FreqTなモデルはF81モデル(Felsenstein, 1981)と呼ばれています。RateAC ≠ RateAG ≠ RateAT ≠ RateCG ≠ RateCT ≠ RateGTであり、かつFreqA ≠ FreqC ≠ FreqG ≠ FreqTなモデル(Tavaré, 1986)は一般時間反転可能(general time-reversibleを略してGTR)モデルと呼ばれています(Posada and Crandall, 1998)。他にも様々なモデルがありますが、全てGTRモデルの下位互換なモデルとなっています。
この後説明する系統樹推定の際には、一般的に無根系統樹を仮定して系統樹推定を行います。そのため、分子進化モデルは時間反転可能なモデルでなくてはなりません(そうでないと尤度が定義できない)。これは、時間反転不能モデルは有根系統樹でしか適用できないのですが、そのためには数値計算の困難さと樹形空間の拡大などの問題があり現実的には難しいためと思われます。
2.1.2 座位間の置換速度不均質性
座位(site)間における置換速度の不均質性(heterogeneity)があることが知られており、これを表すモデルがいくつか提案されています。これらはASRV (among-site rate variation)モデルと呼ばれています。
配列データ内では、置換の滅多にない座位がほとんどであり、置換が頻発する座位は限られています。これにΓ分布を当てはめるものが提案されています(Yang, 1993)。しかし、連続的なΓ分布を当てはめるのは計算量が膨大になるため、Γ分布に基づいて任意の数に座位をカテゴリ分けするモデル(Yang, 1994)が最もよく利用されています。これを + G とか + dG (discrete Gammaの意)などと表記します。カテゴリ分けする数を含めて + dG4 などと表記することもあります。
また、置換の起きない座位(invariable site)と置換が起きる座位(variable site)の2つにカテゴリ分けするモデル( + I と表記)や、 + G と + I を併用したモデルもあります。これらは一定の法則に従って自動的に行われるカテゴリ分けですが、解析者が任意のカテゴリ分け(partitioning)を指定することもできます( + SS (site specific rateの意)と表記)。異なるコドン位置(codon position)や遺伝子座などの置換速度は異なる可能性が高いので、これらがしばしばカテゴリとして指定されます。この場合、単に + SS と表記しても分かりづらいので、 + Codon Position Specific Rate とか + Gene Specific Rate と表記した方が良いでしょう。さらに、これらのカテゴリ内で + G や + I モデルを当てはめることも可能です(ただし、実際には + I モデルを併用できるソフトウェアは存在しません)。 + Codon Position Specific Rate と + G を併用する場合、コドン位置それぞれにΓ分布を当てはめることもできます( + 3 Different Gamma )し、共通のΓ分布を当てはめることも可能です( + 1 Shared/Common Gamma )。同様に、遺伝子座間でもそれぞれにΓ分布を当てはめる場合( + N Different Gamma )と共通のΓ分布を当てはめる場合( + 1 Shared/Common Gamma )があり得ます。隣接する座位間の置換速度の相関を + G モデルに取り入れた + adG (autocorrelated discrete Gammaの意)モデルもあります(Yang, 1995)。
2.1.3 Mixed model
前節では座位(site)間での置換速度不均質性のみを考慮していましたが、塩基置換速度行列および置換速度不均質性の不均質性を考慮することも可能です。つまり、任意の座位のグループ=パーティション(partition)ごとに異なる塩基置換速度行列、異なるASRVモデルを当てはめます。これはmixed modelと呼ばれています。論文によっては区分モデル(partitioned model)と呼んでいることもあります。これに対して、パーティション間に共通の塩基置換速度行列とASRVモデルを当てはめるものは非区分モデル(nonpartitioned model)と呼ばれます。
Mixed modelには大きく分けて3つのモデルが含まれています。1つ目はパーティション間で平均置換速度が等しいと仮定した等速度モデル(partitioned equal mean rate model)で、2つ目はパーティション間での平均置換速度のばらつきを考慮した比例モデル(proportional model)で、もう1つはパーティション間で置換速度の変化が独立している分離モデル(separate model)です。比例モデルでは、パーティション間の置換速度比が系統樹上の全ての枝で共通になっていますが、分離モデルではパーティションごとに全く独立しています。等速度モデルでは枝長パラメータ数は非区分モデルと同じです。比例モデルでは枝長パラメータは増加しませんが、パーティション数-1個のパーティション間の枝長比=置換速度比パラメータの推定が必要になります。分離モデルは枝長パラメータがパーティション数倍の膨大な数となってしまいます。ASRVモデルの中で説明した + SS モデルは、パーティション間で置換速度行列もASRVモデルも共通にしつつ比例モデルを当てはめたのと同じものになります。分離モデルに対してパーティション間の置換速度比が系統樹上の全ての枝で共通という制約を課すと比例モデルになり、比例モデルに対してパーティション間の置換速度比が全て1:1という制約を課せば等速度モデルになる、という関係になっています。
2.2 アミノ酸置換モデル
2.2.1 Empirical model
塩基置換速度行列は4x4の行列でしたが、アミノ酸置換速度行列は20x20の行列となるため、RateXYとFreqXの数は時間反転可能モデルでも 190 + 20 = 210 となり膨大です。そこで、既に系統関係の分かっている分類群間の系統樹において、大量のデータを用いてあらかじめ推定されたRateXY FreqXの値を用いたモデルをアミノ酸置換モデルとして用います。これらは、実際のデータから観測された「経験的な」ものなので、empirical modelと言います。核(Dayhoff et al., 1978; Henikoff and Henikoff, 1992; Jones et al., 1992; Müller and Vingron, 2000; Whelan and Goldman, 2001; Veerassamy et al., 2003; Le and Gascuel, 2008)・ミトコンドリア(Adachi and Hasegawa, 1996; Cao et al., 1998; Abascal et al., 2007)・葉緑体(Adachi et al., 2000)・レトロウィルス(Dimmic et al., 2002; Nickle et al., 2007)のアミノ酸配列用に様々なモデルが提案されています。RateXYは既存のempirical modelの値を用い、アミノ酸頻度FreqXはデータから推定するモデルも + F モデルと呼ばれて広く用いられています。
2.2.2 Empirical mixture model
Empirical modelは所詮empirical modelに過ぎないため、たとえ選択されたとしても手元のデータに本当に適したモデルではない可能性が十分考えられます。だからと言って、20x20の行列のパラメータを推定しながら系統樹推定を行うのは現在のコンピュータの演算能力では困難ですし、近い将来も無理でしょう。そこで、複数のempirical modelを重み付け平均化するモデルが提案されています(Jobb, 2008; Le and Gascuel, 2008, 2010; Le et al., 2012)。主に利用されているのは、Le et al. (2012)のLG4MおよびLG4Xモデルです。平均化の際の重み付けはモデルのパラメータとしてデータから推定する(LG4X)か、あるいは座位間の置換速度不均質性に当てはめる4カテゴリ離散化Γ分布を利用して、異なる速度カテゴリに異なるアミノ酸置換確率行列を当てはめます(LG4M)。有効性が認識されるようになったため、最近になって多くのプログラムに実装されています。また、MrBayesではempirical modelを重み付け平均化するのではなく、model jumpingという方法によって解析中に適用するモデルを変更していきます(Ronquist et al., 2005)。それによって、各モデルの適用された事後確率が得られます。また、モデル平均化(model averaging)によっても類似の効果を得られるかもしれませんが、上述の2つの実装では樹形探索を行いながら平均化パラメータを最適化する、もしくは適用するモデルを切り替えて最適化するというところが大きく異なります。
2.2.3 Mixed model
塩基置換モデルと同様に、ソフトウェアによってはパーティションごとに異なるアミノ酸置換モデルを当てはめるmixed modelを適用することができます。枝長パラメータの扱いに応じて等速度モデル、比例モデルと分離モデルがあるのも同様です。
2.3 より複雑なモデル
これまでのASRVモデルでは、座位間の置換速度不均質性はOTU間では均質であると仮定していました。つまり、置換の高速な座位は系統樹上で変化しないということです。しかし、実際には置換の高速な座位が系統ごとに異なることはあり得ます。そのように制約を緩めたモデルがCovarionモデルです(Tuffley and Steel, 1998)。また、前述のmixed modelでは、パーティションの切り方はa prioriに与えられていなくてはなりませんが、パーティションの切り方自体を自動最適化するモデルが提案されており(Pagel and Meade, 2004)、mixture modelなどと呼ばれています。mixture modelの中でも、いくつのパーティションに切るかをa prioriに指定する必要のないものがCATモデルとしてPhyloBayesというソフトウェアに実装されています(Lartillot and Philippe, 2004)。なお、RAxMLというソフトにもCATモデルというものが実装されていますが、名前が同じなだけで全くの別物です。こちらのCATモデルは、Γ分布を使わずに、座位を任意の数の速度カテゴリに分けて尤度を計算するASRVモデルです。+ G モデルの高速な近似法として用いられています。
また、置換速度行列が系統樹上で変化することを許容するnonhomogeneous model (Blanquart and Lartillot, 2006, 2008)というものも提案されています。これらを突き詰めていくと、最終的には、全ての枝で、全ての座位で、何もかも異なるモデルに至ります。これをno-common mechanisms modelと言います。このモデルでは、置換速度行列はサンプルが1つしかないため最適化が不可能になり、RateXYは全て等しく、FreqXも全て等しいと仮定せざるを得なくなります。全ての座位が別パーティションになるので、ASRVモデルは意味をなしません。そうなると、最節約法による系統樹推定と一致する結果を導くことが証明されています(Tuffley and Steel, 1997)。そのため、no-common mechanisms modelは最節約法で適用されているモデルと言っても差し支えないのかもしれません。
第3章 分子進化モデルの選択
3.1 モデル選択の必要性
モデルに階層性がある(単純なモデルは複雑なモデルの特殊な状況である)場合、尤度計算の際に当てはめるモデルは複雑なものほど当てはまりは良くなりますが、実際にはデータにはノイズが含まれており、ノイズにまでフィットしてしまっても意味が無いどころか有害ですらあります。例えばデータを同じ母集団から採取し直したときに当てはまりが大きく低下してしまうようなら、そのモデルは母集団のパラメータを表しているとは言えません。そこで、パラメータを無制限に増やすのではなく、パラメータ数の増大というコストと尤度の向上という利益のバランスを取る必要が出てきます。それを実現したのがAkaike (1974)によって提案された赤池情報量規準(Akaike information criterionを略してAIC)です。AICは尤度を![]() 、パラメータ数を
、パラメータ数を![]() としたときに以下の式で表されます。
としたときに以下の式で表されます。
| (3.1) |
このAICの値が最小となるモデルが最もバランスの取れたモデルであるということが理論的に導かれています。これを利用して最適なモデルを選択してやればよいわけです。しかし、AICはサンプルサイズが無限大の理想的なデータを前提とした近似によって導かれています。実際のデータはサンプルサイズ無限大ということはあり得ませんので、サンプルサイズが小さいときにAICがパラメータ数の増大コストを過小評価してしまうことを正規分布を仮定して補正したAICcがSugiura (1978)によって提案されています。AICcはサンプルサイズを![]() としたときに以下のように表されます。
としたときに以下のように表されます。
| (3.2) |
ここで、分子進化モデルの選択に対するAICcの適用には「正規分布を仮定して補正した」という点が問題になります。分子進化モデルの誤差構造が正規分布ではないからです。また、![]() が0以下のとき、AICcは算出することができません。これは、そのようなモデルはそのデータには適用すべきでないと考えるべきなのかもしれません。
が0以下のとき、AICcは算出することができません。これは、そのようなモデルはそのデータには適用すべきでないと考えるべきなのかもしれません。
また、「ベイズ的」と言われているBICというものもあります(Schwarz, 1978)。これは以下の式で表されます。
| (3.3) |
このように規準が複数あると、どれを使えばいいのかという話になりますが、筆者は最尤系統樹推定で用いるモデルの選択にはAICかAICc、ベイジアン系統樹推定で用いるモデルの選択にはBICを使うことにしています。AICかAICcかは、それぞれ「サンプルサイズ無限大を前提としている」、「正規分布を仮定している」という問題があり、一長一短があります。筆者は全ての候補モデルでAICcが算出可能、つまり![]() であればAICcを、そうでなければAICを使うことにしています。
であればAICcを、そうでなければAICを使うことにしています。
本来、系統樹推定においては分子進化モデルの選択と系統樹の選択は同時に行われるべきですが、1つの分子進化モデルにおいてさえ、系統樹の選択は大変な労力を要します。そのため、全ての分子進化モデルでそうするのは非現実的です。そこで、とりあえずそれほど悪くはないであろうと考えられる「仮の」系統樹に樹形を固定して(ソフトによっては簡易な樹形探索も行う)、各分子進化モデルにおける最大化対数尤度を計算し、それに基づく情報量規準によって分子進化モデルの選択を行います。その後、選択された分子進化モデルを適用して系統樹の選択を行います。つまり、現状の分子系統樹推定は「多重モデル選択」となっているわけです。reversible jump MCMC (model jumping)などによりいつかは解決されるかもしれませんが、しばらくの間はこの方法が使われ続けることになるでしょう。
この問題があるため、系統樹推定によって最終的に得られた系統樹でも、同じ分子進化モデルが選択されるのかを確認することが望ましいでしょう。もし異なるなら、選択された分子進化モデルを適用して再度系統樹推定を行う必要があります。ただ、何度やってもモデル選択結果と系統樹推定結果が一致しない可能性があります。その場合は複数の暫定最尤系統樹の中でモデル選択に用いた規準の値が最も小さいものを使うか、いずれにおいても共通している部分についてのみ考察するしかないでしょう。
3.2 Kakusan4・Aminosanによる分子進化モデルの選択
Kakusan4・Aminosan (Tanabe, 2011)は、配列データに対して最適な置換モデルを選択し、RAxMLやMrBayes (MrBayes5D)用のモデル設定ファイルを書き出してくれるソフトウェアです。また、モデル選択に必要な尤度の計算はRAxML・PAUP*・baseml・Treefinderのいずれか(AminosanはRAxMLかTreefinderかcodeml)に丸投げして計算させますが、この際に並列化して各ソフトウェアを起動するため、マルチCPUやマルチコアCPUを搭載したコンピュータでは従来のソフトウェアよりも大幅に高速な処理が可能になっています。対応している入力データは、FASTA・NEXUS・PHYLIP・GenBankなどの配列ファイルです。モデル選択に利用できる情報量規準はAIC (Akaike, 1974)・AICc (Sugiura, 1978)・BIC (Schwarz, 1978)となっています。
Kakusan4とAminosanは、以下のような処理を行っています。
-
1.
χ二乗独立性の検定による塩基・アミノ酸頻度の均一性確認
-
2.
固定する仮の系統樹を作成 (指定も可能)
-
–
JC69距離の近隣結合樹 (Kakusan4)
-
–
K83距離の近隣結合樹 (Aminosan)
-
–
-
3.
領域・コドン位置ごとに候補モデルを当てはめて最大化対数尤度を求める
-
4.
領域・コドン位置ごとに情報量規準を算出してモデル選択
-
5.
領域・コドン位置ごとに選択されたモデルを適用した等速度・比例・分離モデルを全領域連結配列に当てはめて最大化対数尤度を求める
-
6.
情報量規準を算出して非区分・等速度・比例・分離モデルからのモデル選択を行う
このように、一旦各領域・各コドン位置ごとのモデル選択を行ってから連結配列での非区分・等速度・比例・分離モデルからのモデル選択をしているため、ここでも多重モデル選択になっています。また、等速度・比例・分離モデルは本来、各領域・各コドン位置に全候補モデルを当てはめる全ての組み合わせからモデル選択すべきですが、計算量的に非現実的なので、各領域・各コドン位置のモデル選択で選ばれたモデルを用いた等速度・比例・分離モデルを当てはめることで妥協しています。
Kakusan4・Aminosanには2つの動作モードがあります。1つ目は誰でも簡単に利用できる(つもりで作った)対話型の動作モードで、2つ目は自動処理に適したコマンドラインから操作する動作モードです。ここでは対話型の動作モードでの操作方法を説明していきます。主にKakusan4を用いて説明していきますが、Aminosanでは異なる点があれば適宜説明を加えていきます。また、現在のところAminosanはEmpirical mixture modelには対応していません。
3.2.1 モデル選択の実行
いずれの環境においても、Kakusan4・Aminosanを起動すると標準で対話モードになります。対話モードでは最初に入力ファイルの名前を質問されます。
Kakusan4 4.0.2012.11.06 ======================================================================= This is a script to select nucleotide substitution model for multi- partitioned data set. Official web site of this script is http://www.fifthdimension.jp/products/kakusan/ . To know script details, see above URL. If you publish your study using Kakusan4, please cite the following. Tanabe AS (2011) "Kakusan4 and Aminosan: two programs for comparing nonpartitioned, proportional, and separate models for combined molecular phylogenetic analyses of multilocus sequence data", Molecular Ecology Resources, vol.11, pp.914-921. Copyright (C) 2006-2012 Akifumi S. Tanabe This program is free software; you can redistribute it and/or modify it under the terms of the GNU General Public License as published by the Free Software Foundation; either version 2 of the License. This program is distributed in the hope that it will be useful, but WITHOUT ANY WARRANTY; without even the implied warranty of MERCHANTABILITY or FITNESS FOR A PARTICULAR PURPOSE. See the GNU General Public License for more details. You should have received a copy of the GNU General Public License along with this program; if not, write to the Free Software Foundation, Inc., 51 Franklin Street, Fifth Floor, Boston, MA 02110-1301 USA. Parsing command line options... No input files are specified. Entering interactive mode. Specified options are ignored. Specify an input file name. Note that you can use wild card.
Windows (Vista以外)・Mac OS X環境では、ここでファイルのアイコンを1つだけこのウィンドウにドロップすると、ファイルのフルパス名が入力されます。Windows Vistaでは、エクスプローラ上でShiftキーを押しながらファイルアイコンを右クリックしてパスとしてコピーをしてから、タイトルバーを右クリックし、編集の中にある貼り付けを行って下さい。
Specify an input file name. Note that you can use wild card. "C:\Users\akifumi\Desktop\SampleData\CYTBnuc_P.fas"↓
そのままEnterキーを押すとファイルがKakusan4・Aminosanに読み込まれます。
"C:\Users\akifumi\Desktop\SampleData\CYTBnuc_P.fas"↓ "C:\Users\akifumi\Desktop\SampleData\CYTBnuc_P.fas" was accepted. Specify an input file name or just press enter to leave input file specification.
複数領域データなどの場合、領域ごとに別のファイルとして用意しておき、この操作を繰り返して全ファイルを読み込ませます。同一の遺伝子配列でも、タンパクコード領域とそうでない領域は必ず別のファイルに分けて下さい。タンパクコードでない領域の中で、イントロンと5’・3’の非翻訳領域は分けるべきかどうかは正直分かりません。場合によるでしょう。なお、タンパクコード領域塩基配列データではファイル名(拡張子は含まない)が必ず_Pで終わるようにして下さい。こうすることで、コドン位置ごとの置換速度の不均質性やコドン位置ごとに異なる塩基置換モデルを当てはめるmixed modelが検討されるようになります。また、複数領域データでは、各ファイルでの配列名が統一されていなくてはなりませんので注意して下さい。入力ファイルの指定の際には*や?といったワイルドカードが使えます。ワイルドカードを用いることで一度に多数のファイルを読み込ませることができます。
また、Aminosanでは、ファイル名が_mtで終わるようにすると、検討対象モデルをmtREV (Adachi and Hasegawa, 1996)・mtMam (Cao et al., 1998)・mtArt (Abascal et al., 2007)・mtZoa (Rota-Stabelli et al., 2009)のみに制限できます。同様に_ncでDayhoff (Dayhoff et al., 1978)・JTT (Jones et al., 1992)・BLOSUM62 (Henikoff and Henikoff, 1992)・VT (Müller and Vingron, 2000)・WAG (Whelan and Goldman, 2001)・PMB (Veerassamy et al., 2003)・LG (Le and Gascuel, 2008)のみに、_cpでcpREV (Adachi et al., 2000)のみに、_rtでrtREV (Dimmic et al., 2002)・HIVb・HIVw (Nickle et al., 2007)のみに検討モデルが絞られます。_モデル名で特定モデルのみに絞ることもできます。ただし、 + F, + G, + I の適用は検討されます。なお、Aminosanが検討するDayhoff・JTTモデルは、Kosiol and Goldman (2005)によるDCMutバージョンと呼ばれる若干の改善が施されたものであることに注意して下さい。
全てのデータファイルを読み込ませたら、何も入力せずにEnterキーを押します。
Specify an input file name or just press enter to leave input file specification. ↓ OK. Input file specification has terminated. Log, result and configuration files will be output to "C:\Users\akifumi\Desktop\ SampleData\CYTBnuc_P.fas.kakusan".
以上のメッセージの通り、最初に与えたファイルの存在するフォルダ内に「最初に与えたファイルのファイル名.kakusan」という名前のフォルダ(Aminosanの場合は末尾はaminosanになります)が作成され、そこに全ての結果が出力されます。続いて、どの系統樹推定ソフトウェア向けのモデル選択を行うのか尋ねてきます。
OUTPUT OPTIONS Which is a target analysis software? (MrBayes/Treefinder/PAUP/PHYML/RAxML) (default: RAxML)
この質問では、選択した系統樹推定ソフトウェア向けのモデル設定ファイルが出力されるように設定されます。タンパクコード領域配列データを入力してTreefinderかRAxMLかMrBayesを選択すると、コドン位置ごとに異なるモデルを当てはめるmixed modelの検討が強制的に有効になります。RAxMLの場合はコドン位置間で共通のモデルを当てはめることも強制的に検討されます。PAUP*またはPHYMLを選択した場合、全領域連結配列を区分せず共通なモデルを当てはめることが強制的に検討されます。これはPAUP*とPHYMLがmixed modelに対応していないからです。この後の質問は、これまでの返答によって内容が変化します。全ての質問に関して説明していきますが、表示されない質問がある場合がありますのでご注意下さい。
次の質問は、コドン位置ごとに異なる塩基置換モデルを当てはめるmixed modelを検討するか否かに関するものです。ただし、タンパクコード領域データを入力していない場合や、強制的に検討される場合には質問が表示されません。また、当然ですがAminosanでもこの質問はされません。
ANALYSIS OPTIONS You input protein coding sequence. Do you want to consider partitioning of codon positions? (y/n) (default: n)
この質問にyと答えてEnterキーを押せば、コドン位置ごとに最適な塩基置換モデルの選択が行われます。
次の質問は、タンパクコード領域において全コドン位置を区分せず共通なモデルの検討を行うか否かに関するものです。ただし、タンパクコード領域データを入力していない場合や、コドン位置ごとに最適なモデル選択を行う設定が有効になっていない場合は表示されません。PAUP*やPHYML用の設定ファイル出力が有効になっている場合にも表示されません。もちろん、Aminosanでもこの質問はされません。
You enabled partitioning of codon positions. Do you want to consider nonpartitioning of codon positions? (y/n) If you say yes, applying nonpartitioned models to all-codon position-concatenate d sequences will be considered on each locus. (default: y)
この質問にnと答えた場合、タンパクコード領域において全コドン位置に共通な非区分モデルの検討は行われません。yと答えるか空欄のままEnterキーを押せば検討されます。
次の質問は、複数領域データを与えている場合に、全領域連結配列に領域を区分しないモデルを当てはめることを検討するか否かに関するものです。複数領域データを与えていない場合や、PAUP*またはPHYML用の設定ファイル出力を有効にしている場合は強制的に有効になるので表示されません。
You input multiple files. Do you want to consider nonpartitioning of loci? (y/n) If you say yes, applying nonpartitioned models to all-loci-concatenated sequence s will be considered. (default: n)
この質問にyと答えてEnterキーを押せば、全領域連結配列に領域を区分しないモデルを当てはめることが検討されますが、nと答えれば検討されません。
次は複数領域データかタンパクコード領域データを与えたときに、全領域連結配列または全コドン位置連結配列における非区分・等速度・比例・分離モデル間の比較を行うかに関する質問です。この質問は複数領域データかタンパクコード領域データを与えていないと表示されません。PAUP*またはPHYML用設定ファイル出力を有効にしている場合も表示されません。また、RAxML用設定ファイル出力を有効にしている場合は比例モデルが検討されないので文言が異なります。
You input multiple files and/or protein coding sequence. Do you want to compare nonpartitioned, partitionedequalmeanrate, proportional, a nd separate models on all-loci concatenated sequences? (y/n) Note that this function needs Treefinder. (default: y)
yと答えるか、何も入力せずにEnterキーを押せば非区分・等速度・比例・分離モデル間の比較が行われます。分離モデルの対数尤度は各領域の対数尤度の和なので簡単に求められますが、等速度モデル・比例モデルの尤度は実際に当てはめて計算しなくてはならないため、計算量が増加します。また、RAxML用設定ファイルの書き出しを有効にしている場合は非区分・等速度・分離モデルだけが検討され、比例モデルは検討されません。これはRAxMLが比例モデルに対応していないためです。この際の尤度の計算はRAxMLで行われます。RAxML以外のソフト用の設定ファイル書き出しを有効にしている場合、等速度モデル・比例モデルの尤度は次の質問の内容にかかわらずTreefinderで計算されます。そして、Treefinder以外で計算した尤度との互換性が厳密にあるのか何とも言えないので、他の尤度計算にTreefinder以外が使われていた場合はTreefinderで尤度を計算し直すためさらに計算量が増加します。また、RAxML・Treefinderが + SS モデルには対応していないためこれは比較対象に含まれていません。つまり、非区分モデルやコドン位置間非区分モデルが選択されなかったとしても、 + SS モデルを検討していないせいである可能性は残ります。
次の質問は、モデル選択に用いる尤度の値をどのプログラムで計算させるかというものです。PAUP*・baseml (Aminosanではcodeml)・Treefinderのいずれかから選びます。この質問はPHYML用設定ファイル書き出しを有効にした場合しか表示されません。RAxML用設定ファイルの書き出しを有効にしている場合はこの質問は出ず、RAxMLで尤度が計算されます。PAUP*用設定ファイル書き出しを有効にしている場合はPAUP*で、MrBayes用設定ファイル書き出しを有効にしている場合はTreefinderで尤度が計算されます。
Which do you want to use the program for likelihood calculation? (baseml/tf/paup) (default: baseml)
basemlと答えれば、basemlが各モデルの尤度最大化に使われます。tfと答えればTreefinderが、paupと答えればPAUP*が用いられることになります。
次の質問は、塩基頻度パラメータを持つモデルにおいて、各塩基頻度パラメータを最適化するか、それともデータから得られる観測値を用いるのかに関するものです。RAxML用設定ファイルを書き出す際にはこの質問は表示されず、パラメータの最適化は行われません。
Do you want to optimize the parameters of base composition? (y/n) (default: n)
nと答えるか、何も入力せずにEnterキーを押すと、最適化が無効になり、データから得た観測値が用いられます。最適化は行われません。yと答えると最適化が行われます。最適化を行うと時間はかかりますがより厳密な解析が行われます。しかし、塩基配列でデータが十分にある場合は最適化の効果はあまりありませんので無効にしても構わないでしょう。アミノ酸配列では形質状態が20もあるため、最適化した方が良いことも多いと思いますが、最適化ができるのはTreefinderで尤度を計算する場合のみです。その場合もTreefinderやMrBayes用の設定ファイルを出力させるときしかこの質問は表示されません。
次に、座位間の置換速度不均質性に対する離散Γ分布の当てはめにおいて、離散化の際のカテゴリ数に関する質問がなされます。この質問もRAxML用設定ファイルを書き出す際には表示されません。RAxML用設定ファイルを書き出す際にはこの値は4に設定されます。
How many rate categories of discrete gamma rate heterogeneity do you want to con sider? (integer) (default: 8)
この質問には、正の整数で答えます。少なくとも4以上の値を入力するようにして下さい。値を大きくするほど尤度は正確になりますが計算時間が延びていきます。
次の質問は、ASRVに + I モデルの当てはめを検討するか否かに関するものです。PAUP*かTreefinderで尤度を計算する設定のときにのみ表示されます。
Do you want to consider invariant model for among-site rate variation? (y/n) (default: n)
デフォルトではnですが、検討させたい場合にはyと答えて下さい。
次の質問は、領域・コドン位置ごとに異なる離散Γ分布の当てはめを行うモデルを検討するか否かに関するものです。なお、この質問は尤度最大化にbasemlを用いる場合にしか表示されません。
Do you want to consider N-GAM model for among-site rate variation? (y/n) Note that this model is very time-consuming. (default: n)
yと答えてEnterキーを押せば、領域・コドン位置ごとに異なる離散Γ分布の当てはめを行うモデルが検討されますが、このモデルの尤度最大化には非常に時間がかかるため注意して下さい。この質問でnと答えても、比例モデルや分離モデルで領域・コドン位置ごとに異なる離散Γ分布を当てはめるモデルは検討されます。
次に、隣接座位間の置換速度自己相関を考慮した離散Γ分布の当てはめを行うモデルを検討するか否かに関する質問がなされます。なお、この質問は尤度最大化にbasemlを用いる場合にしか表示されません。
Do you want to consider autocorrelated discrete gamma model for among-site rate variation? (y/n) Note that this model is very time-consuming. (default: n)
yと答えてEnterキーを押せば、隣接座位間の置換速度自己相関を考慮した離散Γ分布の当てはめを行うモデルが候補モデルに含まれるようになりますが、このモデルの尤度最大化には非常に時間がかかるため注意して下さい。このモデルはデータによっては尤度の改善に大きな効果があるのですが、それ以上に計算に膨大な時間がかかってしまいます。計算時間がそれほど問題にならない小さめのデータセットでは有効にしてもよいでしょう。
次に、領域ごとに異なる樹形を用いて尤度最大化を行うか、共通の樹形を用いるかの質問がなされます。なお、この質問は複数領域のデータを与えた場合にしか表示されません。また、非区分・等速度・比例・分離モデル間の比較を行う場合にはこの質問は表示されず、共通の樹形が使用されます。
Do you want to use different tree topology for parameter optimization on each lo cus? (y/n) (default: n)
この質問にyと答えてEnterキーを押せば、各領域で異なる樹形に基づいてモデル選択が行われますが、nと答えるか、何も入力せずにEnterキーを押した場合は全領域連結配列データから生成された樹形に基づいてモデル選択が行われます。領域間の不調和(incongruence)について検討する場合にはyと答えて下さい。樹形は次の質問で樹形ファイルを指定しない限り、JC69距離(AminosanではK83距離 (Kimura, 1983))に基づいて近隣結合法(neighbor-joining (Saitou and Nei, 1987))によって生成されます。対話モードではなくコマンドラインから用いる場合は他の方法も用いることができます。
次に、尤度最大化に用いる樹形を指定するか否かに関する質問です。
If you want to give tree(s) for parameter optimization, specify an input file na me. Otherwise, just press enter.
もしも尤度最大化に用いる樹形を指定したい場合には、ここでNewickかNEXUS形式の樹形ファイルを指定して下さい。その必要が無ければ、そのままEnterキーを押して下さい。
最後に、同時に起動するプロセスの数に関する質問がなされます。
How many processes do you want to run simultaneously? (integer) (default: 1)
ここで、任意の正の整数を入力してEnterキーを押すと、入力した数だけプロセスが同時起動されます。指定する値は、基本的にはお使いのPCが搭載しているCPU(コア)の数と同数にして下さい。そうすることで、PCの演算能力を最大限に生かすことができます。
以上の全ての質問に答え終えると、以下のような表示がなされます。
All configurations have been completed. Just press enter to run!
心の準備ができたらEnterを押して解析を始めて下さい。解析は場合によっては長時間かかってしまいますが、気長に待っていて下さい。
3.2.2 モデル選択結果を見る
既に述べた通り、最初に与えたファイルの存在するフォルダ内に「最初に与えたファイルのファイル名.kakusan」(Aminosanからの出力では末尾はaminosan)という名前のフォルダ(以降、「出力フォルダ」と呼びます)が作成され、そこに全ての結果が出力されます。下図のように、出力フォルダ内にはChisq・Results・MrBayes・PAUP・PHYML・RAxML・Treefinder・Scores・Logsというフォルダが作成され、さらにその中に様々なファイルが出力されます。なお、指定していないソフトウェアのフォルダは作成されません。
-
•
出力フォルダ
-
–
Chisq
-
*
chisq_partition.txt (各領域のχ二乗検定の結果)
-
*
…
-
*
-
–
Results
-
*
partition_criterion.txt (各領域におけるモデル選択の結果)
-
*
whole_criterion_comparemix.txt (連結配列における非区分・等速度・比例・分離モデルからの選択結果)
-
*
…
-
*
-
–
MrBayes
-
*
partition_criterion_xxx.nex (各領域データと選択されたモデルを適用するコマンドの書かれたNEXUSファイル)
-
*
…
-
*
-
–
PAUP
-
*
partition_criterion.nex (各領域データと選択されたモデルを適用するコマンドの書かれたNEXUSファイル)
-
*
…
-
*
-
–
PHYML
-
*
partition.phy (各領域データ)
-
*
partition_criterion_singlesearch.bat (単一の樹形探索を行うバッチファイル)
-
*
partition_criterion_shotgunsearch.bat (ショットガン樹形探索を行うバッチファイル)
-
*
partition_criterion_bootstrap.bat (ブートストラップ解析を行うバッチファイル)
-
*
partition_criterion_shotgunbootstrap.bat (ショットガンブートストラップ解析を行うバッチファイル)
-
*
…
-
*
-
–
RAxML
-
*
partition.phy (各領域データ)
-
*
partition_criterion_xxx.partition (各領域データに選択されたモデルを適用する設定ファイル)
-
*
partition_criterion_xxx_singlesearch.bat (単一の樹形探索を行うバッチファイル)
-
*
partition_criterion_xxx_shotgunsearch.bat (ショットガン樹形探索を行うバッチファイル)
-
*
partition_criterion_xxx_bootstrap.bat (ブートストラップ解析を行うバッチファイル)
-
*
…
-
*
-
–
Treefinder
-
*
partition_xxx.tf (各領域データ)
-
*
partition_criterion_xxx.model (各領域データに選択されたモデルを適用する設定ファイル)
-
*
partition_criterion_xxx.rates (比例・分離を指定する設定ファイル)
-
*
partition_criterion_comparemodels.tl (非区分・比例・分離モデル間の比較を行うTreefinder Languageスクリプト)
-
*
partition_criterion_xxx_singlesearch.tl (単一の樹形探索を行うTreefinder Languageスクリプト)
-
*
partition_criterion_xxx_shotgunsearch.tl (ショットガン樹形探索を行うTreefinder Languageスクリプト)
-
*
partition_criterion_xxx_bootstrap.tl (ブートストラップ解析を行うTreefinder Languageスクリプト)
-
*
…
-
*
-
–
Scores
-
*
partition_model.txt (各領域における各モデルの最大化対数尤度)
-
*
…
-
*
-
–
Logs (その他のログファイルの出力されるフォルダ)
-
*
…
-
*
-
–
partitionはパーティション名(入力ファイル名)、criterionはモデル選択規準、xxxは非区分・等速度・比例・分離モデルの適用状況を示しています。全領域連結配列はwholeという名前のパーティションとなっています。非Windows環境ではバッチファイルの代わりにシェルスクリプト(拡張子は.batでなく.sh)が作成されます。
χ二乗検定の結果(chisq_partition.txt)の内容は、pgtestcompositionの出力と同じ形式です。p値が0.05以下のとき、OTU間の塩基・アミノ酸組成に有意な差があると考えられます。ただし、このp値が信頼できるデータには条件があり、それを満たしていない場合は末尾にその旨を示すメッセージが出ています。
ここで、塩基・アミノ酸組成がOTU間で均一でなくてはならないパーティションは、適用するモデルによって異なります。後述する非区分・等速度・比例・分離モデルの比較において、遺伝子座間非区分・コドン位置間非区分モデルが選択されていた場合、wholeパーティションの組成だけが均一でなくてはなりません。wholeパーティション全体に対して共通の置換速度行列が適用されるためです。遺伝子座間区分・コドン位置間区分モデルが選択されていた場合、区分したそれぞれのパーティションの組成が均一でなくてはなりません。それぞれで異なる置換速度行列が適用されるためです。つまり、共通の置換速度行列の適用範囲で組成が均一でなくてはならないということです。
もしも塩基・アミノ酸組成に有意な差があったのであれば、データ改変による均一化を検討して下さい。他にも、系統樹上で組成が変化することを許容する不均質モデル(Blanquart and Lartillot, 2006, 2008)の適用を検討するのも良いですが、このモデルを適用できるnhPhyloBayesはかなり解析が遅いので、大規模データに適用するのは難しいと思います。
次に、各領域・コドン位置のモデル選択結果(partition_criterion.txt)をテキストエディタで開いてみると以下のような内容となっています。ただし設定ファイル書き出し対象のソフトによって検討されるモデルは異なります。例えばRAxMLではGTR_Gammaしか検討されません。
| model criterion weight -LnL nparam | SYM_GeneCodonPos1Gamma 5.237279083000e+004 0.98496 2.606139541500e+004 125 | J2ef_GeneCodonPos1Gamma 5.238115467800e+004 0.01504 2.606757733900e+004 123 | SYM_Gamma 5.288409574800e+004 0.00000 2.631904787400e+004 123 | 以下略
左から順に、モデル名、情報量規準の値、Akaike weight、-LnLの値、パラメータ数となっています。GeneCodonPos1Gammaというのは、領域間・コドン位置間に異なる速度を当てはめた上で、領域・コドン位置に共通のΓ分布モデルを当てはめたものです。AICcやBICに基づいたモデル選択の結果では、上記の内容に加えてサンプルサイズの値が記述されています。AICcとBICの計算に用いるサンプルサイズの値は複数考えられるため、それぞれをサンプルサイズに用いてモデル選択を行った結果が出力されています。各出力ファイルで使われているサンプルサイズは以下のようになっています。
-
AICc1・BIC1:
系統樹上での最小塩基置換数(最節約樹長)
-
AICc2・BIC2:
各座位における最小塩基置換数の合計
-
AICc3・BIC3:
各座位における形質状態の合計
-
AICc4・BIC4:
座位数(配列長)
-
AICc5・BIC5:
変異のある座位数
-
AICc6・BIC6:
座位数×配列数
最もよく使われているサンプルサイズはAICc4・BIC4の「座位数」です。
ここで重要なのは、このファイルで最上位になっているモデルが実際の解析で適用されるとは限らないということです。というのも、ここでは比較に用いた候補モデル全ての順位が示されているのであって、たとえ最上位でも解析ソフトの側が対応していなければ適用できないからです。実際に適用されるモデルは、必ず解析ソフトで用いる設定ファイルを直接開いて確認して下さい。
Resultsフォルダに作成されるwhole_criterion_comparemix.txtは、連結配列における非区分・等速度・比例・分離モデル間の比較結果です。内容は以下のようなものです。
| model criterion -LnL nparam | Separate_CodonProportional 1.286036307191e+004 6.373181535953e+003 57 | Proportional_CodonProportional 1.286895735412e+004 6.385478677060e+003 49 | Separate_CodonSeparate 1.288258125450e+004 6.352290627248e+003 89 | Proportional_CodonNonpartitioned 1.401815088065e+004 6.983075440327e+003 26 | Separate_CodonNonpartitioned 1.402149556766e+004 6.976747783830e+003 34 | Nonpartitioned 1.413466486467e+004 7.049332432334e+003 18
このファイル内のモデルはそれぞれ以下のようなものです。
-
•
領域間分離・コドン位置間比例モデル
-
•
領域間比例・コドン位置間比例モデル
-
•
領域間分離・コドン位置間分離モデル
-
•
領域間比例・コドン位置間非区分モデル
-
•
領域間分離・コドン位置間非区分モデル
-
•
非区分モデル
上記は等速度モデル非対応の旧バージョンの出力ファイルなので等速度モデルが登場しませんが、最新版ではPartitionedEqualMeanRateという名前で等速度モデルがリストに現れます。なお、Kakusan4・AminosanはMrBayes (MrBayes5D)とTreefinder用の非区分・等速度・比例・分離モデルを適用する設定ファイルを書き出すことができますが、Kakusan4・Aminosanが複数領域データにおいて実際に行っているのは、既に述べたように「それぞれの領域」での最適モデルの選択と、それぞれの領域で選択されたモデルを用いた非区分・等速度・比例・分離モデル間の比較だけです。これは、領域ごとに当てはめるモデルが多数あるとき、その組み合わせはさらに多数になってしまい、全ての比較を現実的な時間で処理することが不可能だからです。ただし、分離モデルはただそれぞれの領域で最大化した対数尤度を足し合わせたものですので、モデル選択にAICを用いる場合は全ての組み合わせで正攻法で尤度を計算してモデル選択した結果と完全に一致します。AICcやBICは相加的ではないため完全には一致しない可能性があります。
Kakusan4・Aminosanでは、このようにして選択された分離モデルに対して全ての領域・コドン位置で枝長が比例するように制約を課すことで比例モデルの設定ファイルを作成しています。当然、実際には領域・コドン位置間で枝ごとの置換速度のパターンが異なる場合には、部分的に分離モデルを適用し部分的に比例モデルを当てはめたモデルがより良い可能性はありますが、そのような比較は行っていませんし設定ファイルも作成されません。また、非区分・等速度・比例・分離モデル間の比較をTreefinderで行っている場合、Treefinderが対応していないモデルは比較対象に入っていません。比較対象に入っていないモデルがベストである可能性は常に残っていることに注意して下さい。
第4章 最尤系統樹推定
4.1 最尤系統樹推定とは何か
今更ではありますが、そもそも尤度とは「あるモデルが正しいと仮定した状況で手元のデータが得られる確率」のことです。これは、データに対するモデルの当てはまりの良さを表します。ここで、10回のコイントスを行って表が1回、裏が9回出たときの状況を考えましょう。すると、「このコインを使ったコイントスでは表と裏が1:9の比率で出る」というモデルの尤度![]() は以下のようになります。
は以下のようになります。
|
|
|
|
(4.1) | ||
|
|
|
「このコインを使ったコイントスでは表と裏が等確率で出る」というモデルの尤度![]() は以下のようになります。
は以下のようになります。
|
|
|
|
(4.2) | ||
|
|
|
このように、![]() であることから、前者のモデルの方が当てはまりが良いことになります。ただし、前者は「表と裏が1:9の比率」ということをデータから推定していると考えられますので、パラメータが1つありますが、後者にはデータから推定しているパラメータがありませんので、AICは以下のようになります。
であることから、前者のモデルの方が当てはまりが良いことになります。ただし、前者は「表と裏が1:9の比率」ということをデータから推定していると考えられますので、パラメータが1つありますが、後者にはデータから推定しているパラメータがありませんので、AICは以下のようになります。
|
|
|
|
(4.3) | ||
|
|
|
||||
|
|
|
|
(4.4) | ||
|
|
|
ここでも![]() であることから、やはり前者のモデルの方が良いということになります。
であることから、やはり前者のモデルの方が良いということになります。
最尤系統樹推定は、分子進化モデルは固定(パラメータ値はそうでない)にして、最も尤度が高くなるような系統モデル=系統樹を選択するというものです。系統モデルは枝長パラメータと樹形から成りますが、検討すべき樹形数は配列数=系統樹の端点数=OTU (operational taxonomic unit)数に応じて劇的に膨れ上がってしまいます。そのため最尤系統樹推定では、網羅的探索(exhaustive search)は計算時間から見て非現実的です。そこで、ほとんどの場合は発見的探索(heuristic search)を行います。これは、近隣結合法(neighbor-joining (Saitou and Nei, 1987))や段階的配列付加法(stepwise/sequential sequence addition (Swofford and Begle, 1993))などで生成した初期系統樹(initial/starting tree)と、それを枝交換(branch swapping)によって樹形改変(topology rearrangement)してできる系統樹の尤度を計算し、より尤度の高い系統樹が見つかればそれを初期系統樹としてまた同じことを繰り返す、というものです。
4.2 RAxMLによる発見的探索
最尤系統樹推定に用いられるソフトは各種ありますが、ここでは比較的高速なRAxML (Stamatakis, 2006)を用いて説明していきます。ただし、RAxMLは塩基配列データでは置換速度行列はGTRモデルしか使えない上、分離モデルは使えるものの比例モデルはサポートしていません。RAxMLの詳しい使い方はver.7.0.4のマニュアルに書かれていますが内容が古いので、最新版で-hオプションを付けて実行したときに表示されるメッセージを参照するようにして下さい。
Kakusan4・Aminosanで分子進化モデルの選択を行った場合、出力フォルダ下のRAxMLフォルダに
partition_criterion_xxx_singlesearch.bat
というファイルが作成されているはずです。partitionはパーティション名、criterionはモデル選択規準、xxxはmixed modelの適用状況を示しています。連結配列データはwholeという名前のパーティションとなっています。非Windows環境では拡張子は.batではなく.shとなっていますのでご注意下さい。作成されるファイルは入力されたデータが複数領域データか、タンパクコード領域データかによって異なりますが、例えば、
whole_AIC_separate_codonseparate_singlesearch.bat
(AICをモデル選択規準として領域・コドン位置ごとに選ばれたモデルを分離モデルとして連結配列に当てはめて樹形探索を行うバッチファイル)
とか
whole_AIC_codonseparate_singlesearch.bat
(AICをモデル選択規準としてコドン位置ごとに選ばれたモデルを分離モデルとして連結配列に当てはめて樹形探索を行うバッチファイル)
とか
whole_AIC_nonpartitioned_singlesearch.bat
(AICをモデル選択規準として選ばれた非区分モデルを配列全体に当てはめて樹形探索を行うバッチファイル)
といったファイルが出力されているはずです。タンパクコード領域配列では
whole_AIC_nonpartitioned_singlesearch.bat
は
whole_AIC_codonnonpartitioned_singlesearch.bat
という名前で出力されています。ただし、タンパクコード領域において全コドン位置に共通なモデルの検討を行わない設定でKakusan4によるモデル選択を行った場合はこのファイルは出力されません。
上述のことから分かるように、同じパーティションのデータに対して、多くのモデルの当てはめ方があり得ます。上記の例では分離モデルしか挙げていませんが、等速度モデルも利用可能です。どの当てはめ方が最も適しているかはデータによって異なります。どのモデルを当てはめればよいかは第3.2.2節をご覧下さい。
適切なファイルを選んだら、Windows上ではバッチファイルを直接実行すれば解析が行われ、結果がRAxML_*というファイル名で保存されます。Windows以外の環境ではターミナル上で
> sh ファイル名↓
というコマンドを実行すれば解析が走ります。
これらをそのまま実行すると、CPUを1個しか利用しない解析が行われます。このため、より高速に処理したい場合はこのファイルを編集する必要があります。このファイルをテキストエディタで開くと、以下のような内容になっています。
| raxmlHPC -n partition_criterion_xxx_singlesearch -s partition.phy -f d -p 1234 -m GTRGAMMA
これが実行される解析コマンドで、raxmlHPCがコマンド名、それ以降がオプションとなっています。ここで、raxmlHPCをraxmlHPC-PTHREADSに置換し、オプションに-T 8を加えると、CPUは8個使用されます。また、よほど古いCPUでない限り、SSE3という拡張命令に対応しているはずです。この拡張命令を使うと、「複数のデータへの同内容の処理」が1命令で行えるため、適した処理では計算が高速になります。raxmlHPC-PTHREADS-SSE3を実行すれば、この拡張命令が利用されるため、処理が高速化します。ごく最近のCPUでは、AVX命令とかAVX2命令というより強力な拡張命令が利用できます。これらを利用するにはそれぞれraxmlHPC-PTHREADS-AVX・raxmlHPC-PTHREADS-AVX2を実行して下さい。ただし、Windows版は高速化しないかもしれません。
非常にOTUが多いデータセットの場合、1度の発見的探索では尤度の山の第1峰に到達できないことが多くなります。OTUが多いと、それだけ探索空間が広がり、尤度の山も頂上付近がなだらかながらも凹凸がある状態になります。それぞれの峰の頂点は距離があるため容易に移動できません。そのため、1度の探索では第2峰やそれ以下の峰の頂点を尤度の最高点と誤認してしまいます。そこで、初期系統樹を多数用意し、それらからの発見的探索を行なって、全ての探索結果の中から最も尤度の大きいものを採用します。これを行うのが
*_shotgunsearch.bat
という名前のバッチファイルです。このファイルの内容も上記と同様の方法で編集することで高速化できます。標準では初期系統樹の数、つまり発見的探索の回数は10回になっています。-N 10というオプションで指定されています。この値を変えることで探索回数を変更可能です。OTUが多いほどこの値を大きくして下さい。
どちらの解析でも、最尤系統樹はRAxML_bestTree.*という名前で保存されます。
4.3 RAxMLによるブートストラップ解析
樹形の信頼性(credibility)を検討するために、ブートストラップリサンプリング(bootstrap resampling)したデータを用いて系統樹推定を繰り返すことで、各内分枝(internal/interior branch)の再現率を得ます。これが系統樹推定におけるブートストラップ解析です(Felsenstein, 1985)。
Kakusan4でモデル選択を行った場合は、出力フォルダ下のRAxMLフォルダにある
partition_criterion_xxx_bootstrap.bat
を実行して下さい。この解析も、発見的探索と同様に高速化できます。標準では反復数は100に設定されています。-N 100というオプションで指定されていますので、この値を変えることで反復数を変更できます。解析が終わると、各反復での最尤系統樹がRAxML_bootstrap.*というファイルとして保存されています。これだけでは多数の系統樹があるだけで信頼性がわかりません。そこで、Phylogears2のpgsumtreeコマンドを使って、元データの最尤系統樹に見られた各内分枝の出現頻度をカウントし、百分率にして系統樹上の各枝に数値をマッピングします。これは以下のように実行して下さい。
> pgsumtree --mode=MAP --treefile=RAxML_bestTree.* RAxML_bootstrap.* 出力ファイル名↓
出力ファイルはFigTreeで開くことができます。pgsumtreeを使用すると、ブートストラップ解析の結果をより詳細に分析することができます。詳しくは第6.4節をご覧下さい。
第5章 ベイジアン系統樹推定
マルコフ連鎖モンテカルロ法(Markov chain Monte Carlo略してMCMC)を用いたベイジアン系統樹推定(Bayesian phylogenetic inference)は、近年普及してきていますが、まだパラメータ設定や収束(convergence)判定には一定の知識が必要です。ここではMCMCについて簡単に説明した上で、MrBayes (Ronquist and Huelsenbeck, 2003)の改造版であるMrBayes5Dによる系統樹推定と、Tracerを用いた収束判定について説明します。
5.1 メトロポリス・ヘイスティングス法
MrBayes (MrBayes5D)が行うMCMCは、メトロポリス・ヘイスティングス法(Metropolis-Hastings algorithm (Metropolis et al., 1953; Hastings, 1970))と呼ばれるMCMCです。この方法は、以下のような手順でパラメータを最適化していくものです。
-
1.
全てのパラメータの初期値を適当に決定する
-
2.
パラメータを適当に選択する
-
3.
選択されたパラメータを事前分布から導かれる提案分布に従って変更する、ことを提案する
-
4.
パラメータ変更後の尤度を計算する
-
5.
尤度が変更前より良くなっていれば提案を100%受理し、良くなっていなくても一定のルールで受理する
-
6.
2へ戻る
-
7.
以上の処理を継続しつつ、一定の間隔でモデルをサンプリングする
この処理がある程度進むと定常状態(steady state)に入ります。定常状態に入る前のサンプルを捨て(burn-in)、残ったサンプルを事後分布(posterior distribution)からのサンプルと見なして事後確率(posterior probability)を得ます。
5.2 MrBayes5Dによる系統樹推定
MrBayes5Dは現在最もよく利用されているベイジアン系統樹推定用ソフトウェアMrBayesを拡張し、より多くのアミノ酸置換モデルを使えるようにしたものです。塩基配列データの解析にはオリジナルとの違いはありません。計算も高速で多くのモデルに対応しており、MPIによる並列化にも対応しています。以下の説明のほとんどはオリジナルのMrBayesにも適用できます。塩基配列データの解析を行う場合は、本家のMrBayesの方が高速でパラメータの自動チューニングなどにも対応しているため、そちらをお使い下さい。
Kakusan4・Aminosanで分子進化モデルの選択を行った場合、出力フォルダのMrBayesフォルダ内にあるNEXUSファイルを読み込むことで容易に選択されたモデルを適用した解析が可能です。MrBayesフォルダには
partition_criterion_xxx.nex
というファイルが作成されているはずです。partitionはパーティション名、criterionはモデル選択規準、xxxは非区分・比例・分離モデルの適用状況を示しています。全領域連結配列はwholeという名前のパーティションとなっています。作成されるファイルは入力されたデータが複数遺伝子座データか、タンパクコード領域データかによって異なりますが、例えば、
whole_BIC4_proportional_codonproportional.nex
(配列長(座位数)をサンプルサイズとしたBICをモデル選択規準として領域・コドン位置ごとに選ばれたモデルを比例モデルとして連結配列に当てはめる設定を適用するNEXUSファイル)
とか
whole_BIC4_codonproportional.nex
(配列長(座位数)をサンプルサイズとしたBICをモデル選択規準としてコドン位置ごとに選ばれたモデルを比例モデルとして連結配列に当てはめる設定を適用するNEXUSファイル)
とか
whole_BIC4_nonpartitioned.nex
(配列長(座位数)をサンプルサイズとしたBICをモデル選択規準として選ばれたモデルを連結配列に当てはめる設定を適用するNEXUSファイル)
といったファイルが出力されているはずです。タンパクコード領域配列では
whole_BIC4_nonpartitioned.nex
は
whole_BIC4_codonnonpartitioned.nex
という名前で出力されています。ただし、タンパクコード領域において全コドン位置に共通なモデルの検討を行わない設定でKakusan4によるモデル選択を行った場合はこのファイルは出力されません。
このように、同じパーティションのデータに対して、多くのモデルの当てはめ方があり得ます。どの当てはめ方が最も適しているかはデータによって異なります。Kakusan4・Aminosanによる非区分・比例・分離モデルの比較結果を参考にしてどのモデルを適用するか=どのファイルを用いるかを適宜選択して下さい。ただし、尤度計算に用いているTreefinderが対応していないために検討していないモデルもあるので、この結果を過度に信用しない方が良いかもしれません。MrBayes5Dは分離モデルにも対応しているため、分離モデルを適用するNEXUSファイルも出力されていますが、MrBayes5Dはあまり分離モデルを適用した解析を得意としていません(樹形探索範囲が狭くなる)。分離モデルが選択されてしまった場合には、RAxMLによる最尤系統樹推定を推奨します。
MrBayes5Dで以上のファイルを用いてMCMCを実行するには、以下のようにコマンドを実行します。
> mrbayes5d -i partition_criterion_xxx.nex↓ MrBayes > MCMC↓
これでMCMCは走り始めます(NGenオプションで指定しない限り1,000,000ステップ)が、どれだけMCMCを走らせ続けるかが問題です。こちらに関しては次節をお読み下さい。
5.3 Tracerによる収束判定と有効サンプルサイズの推定
MCMCで難しいのは、「収束しているか」と「収束後のサンプル数は十分か」の判断です。これを補助してくれるのがTracerです。MrBayes5DもASDSFという収束判断の参考になる値を出力してくれますが、あまり当てにならないので気にしなくていいでしょう。ASDSFは標準設定では1,000ステップごとに計算されますが、この計算が案外重いので、MCMCコマンドのオプションにDiagnFreq=10000 (10,000ステップごとにASDSFを計算する)などと付けることでこの計算の頻度を変更してやると良いかもしれません。もしくは、MCMCDiagn=Noをオプションとして与えることで最初からASDSFの計算をしないようにしてもいいでしょう。NRuns=1として同時に走らせるMCMCを1つに制限した場合もASDSFは計算されません。
まず、MrBayes5DでMCMCを走らせて、以下のメッセージがでたところで、MrBayes5DはそのままにしてTracerを起動します。MCMC実行時にNGenオプションで指定しない限り1,000,000ステップの時点で表示されるはずです。
MrBayes > MCMC↓ 中略 Continue with analysis? (yes/no):
Tracerが起動したら、FileメニューのImport Trace File...からMrBayes5Dに読み込ませているNEXUSファイルのあるフォルダにあるNEXUSファイル名.run1.pを指定して読み込ませます。同様にNEXUSファイル名.run2.pも読み込ませます。現在のバージョンでは左側ペインへのファイルのドラッグアンドドロップによってファイルを読み込ませることも可能になっています。2つのファイルの読み込みが終わったら、左上のTrace Filesペインで2つのファイル名を選択して反転表示状態にします。複数ファイルを選択するにはCtrlかShiftキーを押しながらファイル名を左クリックして下さい。
ここで読み込ませた2つのファイルは、MrBayes5Dが同時に2つ(標準設定の場合)走らせているMCMCのそれぞれのモデルのパラメータ値などが保存されているログファイルです。この2つのMCMCで、パラメータが定常状態に入っているか、近い値に収束しているかをTracerで図示することで判断しようということです。
さて、この状態で、右側ペインのタブをTraceにして折れ線グラフを表示させます。そして、右下のColour byをTrace Fileに、LegendをNone以外にして下さい。すると、2つのMCMCの折れ線グラフが色分け表示されます(図5.1)。左下のTracesペインで反転表示させるパラメータを変更していくと、右ペインの折れ線グラフもそれに応じて変化していきます。このプロットを見て各パラメータが定常状態(steady state)に入っているかを検討して下さい。もし定常状態に入っていそうもないようであれば、MCMCを継続して下さい。MCMCが中断したら、再度ファイルの読み込みをし直して定常状態に入るまで繰り返して下さい。
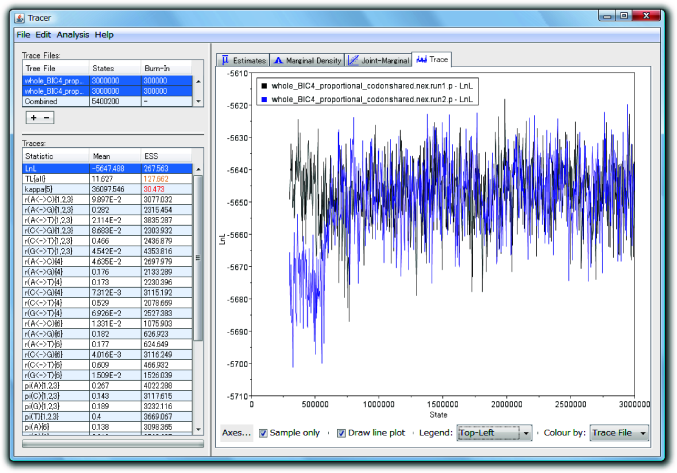
定常状態には入っても、2つのMCMCが異なる局所最適解に収束してしまっている場合、両方のMCMCがより尤度の高い方へと収束するまで解析を続ける必要があります。しかし、あまりに尤度の谷が深いといつまで経っても同じところへ収束しないことがあります。そのような場合、とりあえず尤度の高い方だけが最適解付近に収束していると見なしておき、サンプル数が十分量(後述)の半分程度になるまで解析を続けます。その上で、各種出力ファイルの名前を変更してから何度も同じ解析を実行してやります。そして、2つ以上のMCMCが同じ値に収束しているものの中で、最も尤度の高いものを本当に収束しているものとして結果に採用します。
パラメータが定常状態に入っていると確認できたら、収束後のサンプル数が十分かどうかを検討しましょう。まず、左上のTrace FilesペインのBurn-In (収束前で捨てるサンプル数)を適切な値に設定して下さい。この値は解析開始から定常状態に入るまでのステップ数で指定しますが、ログファイルには第1ステップの結果が入っているため、最初の1,000,000ステップをburn-inするには、この値を1,000,100に設定します。ただし、これは標準の100ステップに1回のサンプリング頻度に設定している(SampleFreq=100)場合で、1,000ステップに1回に設定している場合には1,001,000にします。つまり、burn-inしたいステップ数にSampleFreqの値を加えた値にするということです。ここで、MrBayes5Dの機能上の制約により、解析結果の要約(summarize)するには全ファイルのBurn-Inを等しくしておく必要があります。ただし、MrBayes5Dの要約機能を利用せずに第5.4節の方法で要約するのならばその必要はありません。
全ファイルのBurn-Inを適切に設定できたら、左上Trace FilesペインでCombinedを含む全ての項目が反転表示された状態にして下さい。また、右側ペインのタブをMarginal Densityに設定して事後確率密度を表示させます。そして、右下のColour byをTrace Fileに、LegendをNone以外にして下さい。すると、各MCMCと全MCMCから得られたパラメータの事後確率密度が色分け表示されます(図5.2)。左下のTracesペインで反転表示させるパラメータを変更していくと、右ペインの密度曲線もそれに応じて変化していきます。このプロットを見てMCMC間で同様の密度曲線となっていることを確認して下さい。さらに、左下TracesペインのESS (effective sample size, 有効サンプルサイズ (Kass et al., 1998))の値を見ます。これが全て100、できれば200を超えるようにして下さい。100を下回るようならMCMCをさらに継続してサンプル数を増やす必要があります。
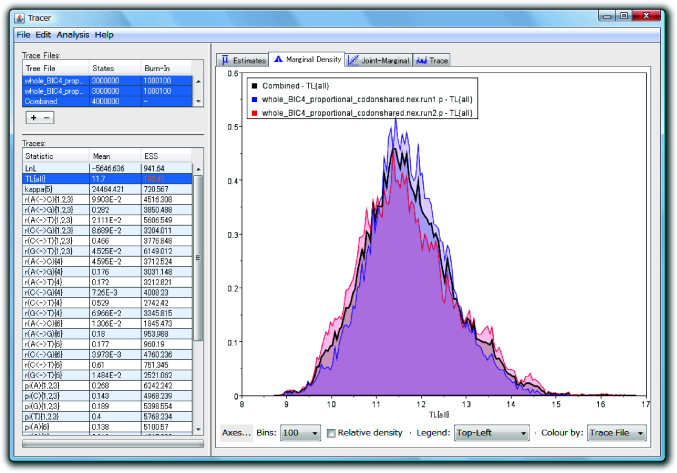
各パラメータの要約統計量を見るには、右側ペインのタブをEstimatesにした状態で、左下Tracesペインのパラメータをクリックして下さい。右側のペイン上部に表示されます。
5.3.1 収束しやすくする・有効サンプルサイズを大きくする方法
MCMCでは、間隔を空けてモデルをサンプルすることで、各サンプルは独立したものとしてみなしています。しかし、独立性が低いとESSの値は小さくなります。ESSは、実際のサンプル数ではなく、サンプル間の独立性を考慮した「実質的なサンプル数」を示しています。提案(proposal)の受理率(acceptance rate)が低かったり、状態交換(state exchange)の成立が少ないと、サンプル間の独立性が下がり、ESSが小さくなります。そのようなMCMCでは、尤度の改善も進みにくいために収束に時間がかかるようになってしまいます。
これを解決するには、2つのアプローチがあります。それは、サンプル間の独立性が低かろうがなんだろうがとにかく長くMCMCを続けてESSを十分な数にするという方法と、サンプル間の独立性を上げてステップ数当たりのESSを大きくする方法です。ESSの不足が少しだけであれば、解析を続けるだけで済む前者の方法を採るのが良いでしょう。しかし、絶望的なまでにステップ数当たりのESSが小さくとてもESSが十分になるまではやっていられないということであれば、設定を変えて解析をやり直すしかありません。
受理率は、MCMCを停止したときに表示されますのでその値を見てどの提案の受理率が低いのかを確認します。以下のように表示されているはずです。
Acceptance rates for the moves in the "cold" chain: With prob. Chain accepted changes to 1.23 % param. 1 (state frequencies) with Dirichlet proposal 以下略
この値が低く、ESSがとても確保できないものをメモしておき、Propsコマンドを使って設定を変更します。MCMCの最中でも、Tracerでパラメータ値の変遷を表示させることでパラメータの最適化の進行とかき乱れの良さを確認できます。横軸をステップ数、縦軸をパラメータ値とするプロットにおいて、上下に激しく乱れておらず矩形波になっているものがあれば、そのパラメータの最適化が進んでいないか、かき乱れが良くないと考えられます。受理率が高くても、提案頻度が低すぎて矩形波になることもあります。Tracerによる確認方法ではそれを発見可能ですので、こちらの方法の方がおすすめです。Propsコマンドは、以下のように用います。
MrBayes > Props↓ 中略 Select a parameter to change (1 - 36; 0 to exit; 37 to zero all proposal rates): 26↓ # 変更するパラメータを選択 Proposal 26: Change (rate multiplier) with Dirichlet proposal # 提案頻度は変更しないときは空欄のままEnter New proposal rate (<return> to keep old = 1.000):↓ # 提案の過激さを変更する New Dirichlet parameter (<return> to keep old = 500.000): 50000↓ 中略 # 設定変更を終了する Select a parameter to change (1 - 36; 0 to exit; 37 to zero all proposal rates): 0↓
proposal rateはそのパラメータの変更が提案される相対頻度で、値を大きくするとより変更の提案される頻度が高くなります。提案の受理率が高くても提案頻度が低い場合はこの値を変更します。提案の受理率が低すぎるのであれば、提案頻度を上げるよりももう一方の値(提案の過激さを決定する)を変更した方が良いでしょう。この値は、大きいほど過激な提案がされたり、逆に小さいほど過激な提案がされたりとパラメータによって意味が変わりますので、MrBayesのマニュアルを見て大きくするか小さくするかを考えて下さい。多くの場合、提案が過激すぎて十中八九受理されないパラメータ値が提案されてしまっていることが多いでしょうから、提案を穏当にする方向へ値を変更すると良いでしょう。設定後にMCMCコマンドでMCMCを走らせ始めることで設定の適用されたMCMCを走らせることができます。MCMCの途中で変更することはできません。
複数領域データやタンパクコード領域データでは、比例・分離モデルやコドン位置ごとに異なる置換速度を当てはめるモデルが適用されていることが多いと思います。しかし、MrBayes5Dでは比例モデルを適用しているときにパーティションごとの置換速度パラメータ(rate multiplier)を提案するDirichlet proposalの受理率が異常に低くなることがしばしばあります。デフォルトではDirichlet parameter (提案の過激さを示す。小さいほど過激な提案がなされる)は1000 (純正のMrBayesでは500)に設定されていますが、もっと大きな値にして提案を穏当にしてやることで改善できることがあります。比例関係にあるパーティション数が多い時にこの問題が起きやすいようです。また逆に、デフォルト値では提案が穏当すぎて最適化が進まず、収束に時間がかかってしまうこともあり得ます。
MrBayes5Dは、同時に2つのMCMCを走らせていると書きましたが、その2つのMCMCのそれぞれはさらに4つのMCMCを同時に走らせています。4つの中には乱数の乱れ(temperature)が大きい=より過激な提案がなされる高温系列(heated chain)が3つ(temperatureは異なる)と、乱数の乱れが最も小さい低温系列(cold chain)が1つあり、MCMCからのサンプリングはこの低温系列から行われています。各系列間ではモデル状態の交換が一定の頻度で試行されます。これをMetropolis-coupled MCMC、略してMC![]() と言います。パラレル・テンパリング法と呼ぶこともあります。こうすることで、より早く収束し、かき乱れが良くなるため、少ないステップ数で大きなESSを得られます。状態交換(state exchange)の試行が成立するかどうかはMetropolis et al. (1953)およびHastings (1970)のルールに従って決定されます。これは前述のパラメータ変更に関しても同じです。
と言います。パラレル・テンパリング法と呼ぶこともあります。こうすることで、より早く収束し、かき乱れが良くなるため、少ないステップ数で大きなESSを得られます。状態交換(state exchange)の試行が成立するかどうかはMetropolis et al. (1953)およびHastings (1970)のルールに従って決定されます。これは前述のパラメータ変更に関しても同じです。
MCMCを停止すると表示されるメッセージの中に以下のようなものがあります。
Chain swap information for run 1: 1 2 3 4 -------------------------- 1 | 0.07 0.01 0.01 2 | 10293 0.04 0.03 3 | 9928 10392 0.05 4 | 10394 9827 9919 中略 Upper diagonal: Proportion of successful state exchanges between chains Lower diagonal: Number of attempted state exchanges between chains
これが状態交換試行の回数と交換の成立率です。1が低温系列で、2~4は順に温度が高くなっていく高温系列を示しています。温度の隣接した系列間の交換成立率が上記のように低い場合、温度の間隔(標準では0.2)を狭くしてやることで交換成立をしやすくすることで改善できる可能性があります。これは以下のようなコマンドで設定可能です。
MrBayes > MCMCP Temp=0.15↓
この設定後にMCMCコマンドでMCMCを走らせ始めると、上記の設定が適用されたMCMCになります。
5.4 解析結果の要約
MCMCを停止したら、そのままでは何らかの意味を見出すのは難しいので、その結果を要約する必要があります。まず初めに、burn-in (収束前で捨てるサンプル数)を決めます。前述したTracerとは違い、ここでのburn-inは解析開始からのステップ数ではなく、サンプル数です。つまり、100ステップに1回サンプルする設定(標準設定)で最初の1,000,000ステップを捨てるには、burn-inは10,001にします(MrBayes5Dは初期状態=第1ステップを保存するため1多くなる)。最初の何ステップを捨てるべきかを判断する方法は第5.3節で説明しています。次に、MrBayes5Dが生成する.tファイルから要約を行います。MrBayes5DのSumTコマンドを用いる方法と、Phylogears2を用いる方法があります。後者は複数のMCMCでburn-inの値が異なる場合にも対応できます。
SumTコマンドを用いて要約を行う場合、MrBayes5DにNEXUSデータファイルを読み込ませた後、以下のようにコマンドを実行します。integerにはburn-inするサンプル数を入力します。
MrBayes > SumT BurnIn=integer↓
これで.conファイルと.partsファイルが作成されます。.conはMCMCからのサンプル系統樹群から生成された多数決合意樹で、枝長は互換性のある系統樹群での平均値です。内分枝(internal/interior branch)の出現頻度はこのファイルにも書かれていますが、.partsをテキストエディタで開くと、対立する内分枝も含めた支持率が書かれています。
Phylogears2を用いる場合、まずはPhylogears2のpgsplicetreeで必要な系統樹だけを取り出します。以下のようにコマンドを実行します。
> pgsplicetree from-to 入力ファイル 出力ファイル↓
from-toには取り出す系統樹の番号を入力します。10002-.などと指定します。これは、10,002本目の系統樹から最後の系統樹までを出力ファイルに取り出すという意味です。これで、最初の10,001本の系統樹はburn-inされることになります。-500-.と指定すれば、最後から500本目の系統樹から最後の系統樹までを出力ファイルに取り出すことができます。複数の.tファイルがある場合(標準設定では2つできます)、以上の処理を全ての.tファイルに対して行った後、pgjointreeで出力したファイルを結合します。以下のようにコマンドを実行して下さい。
> pgjointree 入力ファイル1 入力ファイル2 出力ファイル↓
入力ファイル名は3つ以上指定することも可能です。このファイルをpgsumtreeに与えて出力を得ます。pgsumtreeの使い方は第6.4節をご覧下さい。
その他の各種パラメータの要約は第5.3節をご参照下さい。
5.5 MrBayes5D MPI版による並列計算
インストール方法のところで述べたように、MrBayes5DはMPIによる並列化版(Altekar et al., 2004)があり、これを用いることで大規模な解析を高速に行うことができます。~/にmrbayes5d-mpiとして実行ファイルがあるとすると、起動するには以下のようにコマンドを実行します。
> mpirun -np 利用するCPU数 ~/mrbayes5d-mpi -i NEXUSファイル名↓
なお、MPIフレームワークとしてLAM/MPIをインストールした場合はmpirunで起動する前にlamboot -vを実行しておく必要があります。解析後はlamhaltを実行しておきます。起動後は通常版と同様に扱うことができますが、Propsコマンドによる提案に関する各種パラメータの変更を正常に行うことができません。そのため、これらのパラメータを変更したい場合は、ソースコードに書かれているパラメータを直接書き換え、そのソースから作成した実行ファイルを用いる必要があります。当該箇所はmcmc.cのSetUpMoveTypes関数にあります。
MrBayes5Dでは、標準では4系列(NChains)![]() 2セット(NRuns)で合計8系列のMCMCが実行されます。この状態では最大で8つまでしかCPUを用いることができません。1つの系列に複数のCPUを割り当てることができないためです。大量のCPUがあっても、1つの系列当たりの解析を高速化することはできません。系列数を増加させて温度間隔を狭くすることで系列間の状態交換試行が成立しやすくすることはできますが、劇的に高速化したりはしません。逆に1ステップ当たりの状態交換試行数(NSwaps)を増やさないと、交換の絶対数が減ってしまって系列間の混合具合が悪くなってしまいます。NRunsを増やしても、必要なサンプル数を確保するためのステップ数を小さくすることはできますが、計算そのものを高速化はできません。大量のCPUを用いた高速化が必要な場合は、ExaBayes (Aberer et al., 2014)をご利用下さい。
2セット(NRuns)で合計8系列のMCMCが実行されます。この状態では最大で8つまでしかCPUを用いることができません。1つの系列に複数のCPUを割り当てることができないためです。大量のCPUがあっても、1つの系列当たりの解析を高速化することはできません。系列数を増加させて温度間隔を狭くすることで系列間の状態交換試行が成立しやすくすることはできますが、劇的に高速化したりはしません。逆に1ステップ当たりの状態交換試行数(NSwaps)を増やさないと、交換の絶対数が減ってしまって系列間の混合具合が悪くなってしまいます。NRunsを増やしても、必要なサンプル数を確保するためのステップ数を小さくすることはできますが、計算そのものを高速化はできません。大量のCPUを用いた高速化が必要な場合は、ExaBayes (Aberer et al., 2014)をご利用下さい。
第6章 系統樹の編集・統計と可視化
6.1 クレード・単系統・側系統・多系統・祖先的・派生的
ここではよく使う用語の定義を説明しておきます。
まず、クレード(clade)についてです。クレードとは、系統樹上で複数のOTUが所属する部分系統樹のことです。ただし、有根系統樹と無根系統樹ではやや意味が異なります。無根系統樹では、ある内分枝(internal/interior branch)の一方の端点に接続されている部分系統樹をクレードと言いますが、有根系統樹では内分枝の根から遠い側の端点に接続されている部分系統樹を指します。つまり、有根系統樹上のクレードが根点を含むことはありません。
次に、単系統(monophyly)・側系統(paraphyly)・多系統(polyphyly)です。有根系統樹上で、クレードを形成する分類群を単系統群(monophyletic group)と呼びます。これに対して、メンバーを全て含んでいる最小のクレード内にメンバーでない単系統群を含み、かつ全メンバーに共通の祖先とそこから各OTUまでの枝に当たる生物がその分類群に分類されるような分類群を側系統群(paraphyletic group)と言います。例えば魚類や両生類、爬虫類は側系統群です。メンバーを全て含んでいる最小のクレード内にメンバーでない単系統群を含み、かつ全メンバーに共通の祖先とそこから各OTUまでの枝に当たる生物のいずれかがその分類群に分類されないのが多系統群です。この定義では、共通祖先と共通祖先から各OTUまでの枝に当たる生物の形質状態が問題になり、特定できない状況ではこれらの言葉は使わないようにすべきだと思います。
念のため図6.1のような有根系統樹の場合を考えましょう。この系統樹では、(TaxonA, TaxonB)・(TaxonC, TaxonD)・(TaxonA, TaxonB, TaxonC, TaxonD)は単系統群です。(TaxonA, TaxonB, TaxonC)・(TaxonA, TaxonB, TaxonD)・(TaxonA, TaxonC, TaxonD)・(TaxonB, TaxonC, TaxonD)は、根点に当たる共通祖先と根点から各OTUまでの枝が同一分類群に分類されるなら側系統群です。(TaxonA, TaxonC)・(TaxonA, TaxonD)・(TaxonB, TaxonC)・(TaxonB, TaxonD)は、共通祖先(根点)か根点から各OTUまでの枝のどこかが同一分類群でないなら多系統で、同一分類群であると言えるなら側系統群です。

最後に、祖先的(ancestral/plesiomorphic)・派生的(derived/apomorphic)という言葉に関する注意点です。この言葉は二つの意味で使われています。一つは特定の形質(やそれを有するOTU・単系統群)を指して実際により古くからあるものを祖先的、新しいものを派生的と言っている場合です。もう一つは、単に有根系統樹上でより根に近い(間にある分岐数が少ない)ものを祖先的、遠いものを派生的と言っている場合があります。この二つを混同しないように注意が必要です。というのも、根に近い単系統群の形質が祖先的であるとは限らないからです。
6.2 系統樹ファイルの形式と相互変換
系統樹のファイル形式は主にPHYLIP/Newick形式とNEXUS形式があります。PHYLIP/Newick形式は以下のようなものです。
| 3 | (TaxonA:0.1,TaxonB:0.1,(TaxonC:0.1,TaxonD:0.1):0.1); | (TaxonA:0.1,TaxonC:0.1,(TaxonB:0.1,TaxonD:0.1):0.1); | (TaxonA:0.1,TaxonD:0.1,(TaxonB:0.1,TaxonC:0.1):0.1);
最初の行はファイル中の系統樹の本数を示していますが、これは省略されていることもあります。コロン(:)の後ろの数字は枝長を示しています。PHYLIP形式は、OTU名に使用できる文字数が10文字までである点がNewick形式との違いです。これに対して、NEXUS形式は以下のようになっています。
| #NEXUS | | Begin Trees; | tree tree_1 = [&U] (TaxonA:0.1,TaxonB:0.1,(TaxonC:0.1,TaxonD:0.1):0.1); | tree tree_2 = [&U] (TaxonA:0.1,TaxonC:0.1,(TaxonB:0.1,TaxonD:0.1):0.1); | tree tree_3 = [&U] (TaxonA:0.1,TaxonD:0.1,(TaxonB:0.1,TaxonC:0.1):0.1); | End;
系統樹部分の体裁はほとんど同じですが、Treesブロック内に書かれています。[&U]は、系統樹が無根系統樹であることを示しています。有根系統樹では[&R]になります。この記述は省略可能です。また、下記のようにTranslateコマンドを用いて系統樹内のOTU名を数字に置き換えているものもあります。
| #NEXUS | | Begin Trees; | Translate | 1 TaxonA, | 2 TaxonB, | 3 TaxonC, | 4 TaxonD; | tree tree_1 = [&U] (1:0.1,2:0.1,(3:0.1,4:0.1):0.1); | tree tree_2 = [&U] (1:0.1,3:0.1,(2:0.1,4:0.1):0.1); | tree tree_3 = [&U] (1:0.1,4:0.1,(2:0.1,3:0.1):0.1); | End;
大量の系統樹を1ファイルに保存するときにはこちらの形式の方が容量は小さくなるでしょう。
6.2.1 Phylogears2による変換
Phylogears2には、系統樹ファイル形式を変換することができるpgconvtreeコマンドが含まれています。PHYLIP/Newick・NEXUSに加えてTreefinderのTL Report形式を読み込み、Newick/PHYLIPかNEXUS形式へ書き出すことができます。使い方は下記のようになります。
> pgconvtree --output=Newick 入力ファイル 出力ファイル↓ > pgconvtree --output=NEXUS 入力ファイル 出力ファイル↓
Translateコマンドを使用しているNEXUS形式を読み込むことはできますが、書き出すことはできませんので注意して下さい。
6.3 系統樹の有根化と樹形の変形
6.3.1 Phylogears2による有根化と樹形改変
現在開発中です。
6.4 内分枝出現頻度の分析
そもそも系統樹は複数の系統仮説の集合体です。 たとえば、図6.2aの最尤系統樹には図6.2b, cのような系統仮説が含まれています。 つまり図6.2b, cの系統仮説は互いに矛盾せず同時に成立し得る=互換性がある、と言えます。 また、系統樹そのものもまた多数の互換性のある系統仮説が同時に成立するという系統仮説です。 系統仮説の実体は系統樹上に現れる内分枝(他の枝とのみ接している枝)なので、図6.2b–eのように系統仮説もまた系統樹として表現することができます。
多数決合意樹を見れば、最も多く出現する系統仮説=内分枝は分かりますが、それらと矛盾する内分枝の再現率は分かりません。そこで、Phylogears2に含まれているpgsumtreeを用いることで、ブートストラップ解析やMCMCで現れた全ての内分枝の出現頻度を得ることができます。
まず、コマンドプロンプトやターミナルを起動して、ブートストラップ解析の各反復から得られた系統樹(第4.3節に従って解析した場合はRAxML_bootstrap.*というファイル)またはMCMCの結果が保存してあるフォルダに移動します。そして、以下のようにコマンドを実行します。なお、--mode=CONSENSEにすれば、多数決合意樹を出力させることもできます。
> pgsumtree --mode=ALL 入力ファイル 出力ファイル↓
解析結果は入力ファイルと同じ形式の系統樹ファイルとなっています。仮にNewick形式のファイルを入力ファイルとして与えて開いたとすると、下記のようになっているはずです。この例は16OTUのデータで100反復のブートストラップ解析結果をpgsumtreeで解析したものです。
| [majorhypothesis_1] ((TaxonA,TaxonB,TaxonC,TaxonD,TaxonE,TaxonF,TaxonG,TaxonH,TaxonI,TaxonJ,TaxonK,TaxonL,TaxonM,TaxonN)100.0,(TaxonO,TaxonP)); | [majorhypothesis_2] ((TaxonA,TaxonO,TaxonP,TaxonB,TaxonC,TaxonD,TaxonE,TaxonF,TaxonG,TaxonH,TaxonI,TaxonJ,TaxonM,TaxonN)100.0,(TaxonK,TaxonL)); | [majorhypothesis_3] ((TaxonA,TaxonB,TaxonC,TaxonD,TaxonE,TaxonF,TaxonH,TaxonI,TaxonJ,TaxonK,TaxonL,TaxonM)100.0,(TaxonO,TaxonP,TaxonG,TaxonN)); | [majorhypothesis_4] ((TaxonA,TaxonO,TaxonP,TaxonB,TaxonE,TaxonF,TaxonG,TaxonH,TaxonI,TaxonJ,TaxonK,TaxonL,TaxonM,TaxonN)100.0,(TaxonC,TaxonD)); | [majorhypothesis_5] ((TaxonA,TaxonO,TaxonP,TaxonC,TaxonD,TaxonF,TaxonG,TaxonH,TaxonI,TaxonJ,TaxonK,TaxonL,TaxonM,TaxonN)98.0,(TaxonB,TaxonE)); | [majorhypothesis_6] ((TaxonA,TaxonO,TaxonP,TaxonB,TaxonC,TaxonD,TaxonE,TaxonF,TaxonH,TaxonI,TaxonJ,TaxonK,TaxonL,TaxonM)85.0,(TaxonG,TaxonN)); | 略 | [minorhypothesis_1] ((TaxonA,TaxonO,TaxonP,TaxonB,TaxonE,TaxonF,TaxonG,TaxonH,TaxonJ,TaxonK,TaxonL,TaxonM,TaxonN)25.0,(TaxonC,TaxonD,TaxonI)); | [minorhypothesis_2] ((TaxonA,TaxonB,TaxonC,TaxonD,TaxonE,TaxonF,TaxonH,TaxonI,TaxonJ,TaxonK,TaxonL)21.0,(TaxonO,TaxonP,TaxonG,TaxonM,TaxonN)); | [minorhypothesis_3] ((TaxonA,TaxonB,TaxonC,TaxonD,TaxonE,TaxonF,TaxonH,TaxonI,TaxonK,TaxonL,TaxonM)17.0,(TaxonO,TaxonP,TaxonG,TaxonJ,TaxonN)); | [minorhypothesis_4] ((TaxonA,TaxonH,TaxonJ)15.0,(TaxonO,TaxonP,TaxonB,TaxonC,TaxonD,TaxonE,TaxonF,TaxonG,TaxonI,TaxonK,TaxonL,TaxonM,TaxonN)); | [minorhypothesis_5] ((TaxonA,TaxonO,TaxonP,TaxonB,TaxonE,TaxonF,TaxonG,TaxonH,TaxonJ,TaxonK,TaxonL,TaxonN)14.0,(TaxonC,TaxonD,TaxonI,TaxonM)); | [minorhypothesis_6] ((TaxonA,TaxonC,TaxonD,TaxonM)12.0,(TaxonO,TaxonP,TaxonB,TaxonE,TaxonF,TaxonG,TaxonH,TaxonI,TaxonJ,TaxonK,TaxonL,TaxonN)); | 略
majorhypothesisは多数決合意樹に出力された内分枝を表す系統樹で、全て互いに互換性があります。minorhypothesisは多数決合意樹とは矛盾する内分枝=非互換な仮説を表す系統樹で、majorhypothesisのいずれか1つ以上の系統仮説と非互換な仮説群です。minorhypothesisの仮説間は互換性があるものも無いものも混じっています。いずれも系統樹にも出現頻度が含まれています。85%の確率で出現したmajorhypothesis_6という系統仮説は、TaxonGとTaxonNからなるクレードと、それ以外のOTUからなるクレードとを隔てる内分枝であることを表しています。これと非互換な系統仮説を探すには、minorhypothesisの中から探せばいいわけです。ただ、目視で探すのは面倒なので、それよりは多少楽で確実な方法を用意してあります。まずはpgsplicetreeコマンドを用いてmajorhypothesis_6だけを別ファイル(仮にmajorhypothesis_6.nwkとする)に取り出します。
> pgsplicetree 6 入力ファイル majorhypothesis_6.nwk↓
その上で、以下のようにしてこの出力ファイル内の系統樹と非互換な系統仮説をブートストラップ解析やMCMCの結果から探し出します。
> pgsumtree --mode=ALLi --treefile=majorhypothesis_6.nwk 入力ファイル 出力ファイル↓
出力結果をテキストエディタで開くと以下のようになっています。
| [majorincompatible_1_of_tree_1] ((TaxonA,TaxonB,TaxonC,TaxonD,TaxonE,TaxonF,TaxonH,TaxonI,TaxonJ,TaxonK,TaxonL,TaxonM,TaxonN)8.0,(TaxonO,TaxonP,TaxonG)); | [minorincompatible_1_of_tree_1] ((TaxonA,TaxonB,TaxonC,TaxonD,TaxonE,TaxonF,TaxonG,TaxonH,TaxonI,TaxonJ,TaxonK,TaxonL,TaxonM)7.0,(TaxonO,TaxonP,TaxonN));
majorincompatible_N_of_tree_Kは、入力ファイル内で見られる系統仮説の中で、--treefileオプションで指定した系統樹ファイルのK番目の系統樹と非互換なもので、かつN番目に出現頻度の高いものです。Nが2以上のものもあるかもしれませんが、これはN=1の系統仮説と互換性があるということです。minorincompatibleはmajorincompatibleのどれか1つ以上の仮説と非互換な仮説であることを表しています。majorincompatibleの仮説間では互換性がありますが、minorincompatibleの仮説間では互換性があったり無かったりします。majorincompatible_1は非互換な仮説の中で出現頻度最大なので第2位の仮説と言えるでしょう。minorincompatible_1は第3位の仮説と考えられます。第4位以下の仮説を探すには、1位から3位までの全ての仮説のいずれとも非互換な仮説を探さなくてはなりませんが、まだその方法は用意していません。出現頻度はブートストラップ解析の反復数・MCMCのサンプル数が小さいとかなり変動しますので、第2位の仮説が本当に第2位かどうかはよく検討する必要があります。
第7章 仮説検定
第6.4節で述べたようにして非互換な系統仮説を探すことができます。また、過去の論文から非互換な系統仮説を得られることもあるでしょう。いずれかの仮説を厳密に棄却できるかどうかを検討するには、各系統仮説を制約として課した系統樹推定の結果を比較します。ここではRAxMLによる制約付き最尤系統樹推定とMrBayes5Dを用いた制約付きベイジアン系統樹推定の方法と、制約付き最尤系統樹推定の結果に基づいたKH・SH・AU検定、Bayes factorによる仮説比較について説明します。
7.1 RAxMLによる樹形制約付き最尤系統樹推定
RAxMLで樹形制約(topological constraint)を課した系統樹推定を行うには、まず制約となる系統樹を作成する必要があります。例えば、TaxonA~TaxonEの5 OTUのデータでTaxonAとTaxonBの単系統性(monophyly)を制約として課す場合、以下のような系統樹ファイルを用意します。
| ((TaxonA,TaxonB),TaxonC,TaxonD,TaxonE);
以下のようにしても無根系統樹として見れば意味は同じです。
| ((TaxonA,TaxonB),(TaxonC,TaxonD,TaxonE));
TaxonAとTaxonBの単系統性だけでなく、さらにTaxonAとTaxonBとTaxonCの単系統性も課すには、以下のようなファイルにします。
| (((TaxonA,TaxonB),TaxonC),TaxonD,TaxonE);
このように、「特定の系統仮説を満たす」樹形制約を正の制約(positive constraint)と言います。正の制約下の系統樹推定では、その系統仮説と互換性のある系統樹の中でベストな系統樹を探索することになります。「特定の系統仮説を満たさない」という制約もあり、これを負の制約(negative constraint)と呼びます。負の制約下の系統樹推定では、その系統仮説と互換性の無い系統樹の中でベストな系統樹を探索することになります。RAxMLは負の制約に対応していないため、単系統「でない」という制約を課すことができません。しかし、ブートストラップ解析結果から得られる内分枝出現頻度を見れば、その負の制約下で最も尤度の高い樹形を含む正の制約=第2位の系統仮説が推定できます(必ずこうなるわけではありませんが)ので、それを課した樹形探索を行うことで対処することができます。
制約を表す系統樹ファイルが用意できたら、
partition_criterion_xxx_shotgunsearch.bat
をテキストエディタで開いて編集し、オプションに-g 樹形制約の系統樹ファイルを加えて下さい。これで制約を課した最尤系統樹推定が行われます。また、-nオプションの文字列も変更して下さい。このオプションは出力ファイルの拡張子を指定するものです。例えば-n constrainedMLとした場合、制約付き最尤系統樹はRAxML_bestTree.constrainedMLという名前になります。編集したら念のため別名で保存し、実行して下さい。
7.2 CONSELによる仮説検定
複数の系統仮説を比較したいとき、それぞれの仮説を制約として課した最尤系統樹推定によって得られた制約下の最尤系統樹を比較してやることで、どの仮説が他の仮説より良いか、それは有意な違いかを調べることができます。また、特定の単系統性を検証したいときは、その単系統性の制約下の最尤系統樹と、その単系統性とは矛盾する仮説、即ち負の制約下の最尤系統樹とを比較してやればよいでしょう。ここではブートストラップリサンプリングを応用した検定法の実行方法について説明します。
7.2.1 KH・SH・AU検定
ブートストラップリサンプリングによって、複数の系統樹間で尤度の差が有意と言えるのか否かを調べる方法がKishino-Hasegawa検定(KH test)です(Kishino and Hasegawa, 1989)。しかし、この方法では3つ以上の系統樹を比較する場合に多重検定となってしまい第1種の過誤(有意な差は無いのに誤検出する)が増大してしまうため、その抑制を行う補正を加えたのがShimodaira-Hasegawa検定(SH test)です(Shimodaira and Hasegawa, 1999)。ただし、この方法では逆に第2種の過誤(有意な差があるのに検出できない)が増大してしまいます。そこで、近似的に不偏な検定(approximately unbiased (AU) test)は、マルチスケールブートストラップ法を用いてさらに高度な補正を行うことでこれをある程度解決しています(Shimodaira, 2002)。
CONSELでこれらの検定を行うには、あらかじめ比較する系統樹を最尤法で推定しておきます。これまでの説明の通りに解析を行っていれば、RAxML_bestTree.*というファイルができているはずです。まずはこれらをpgjointreeで結合します。
> pgjointree 入力ファイル1 入力ファイル2 出力ファイル↓
比較したい系統樹が3つ以上ある場合は3つ以上入力ファイルを指定して下さい。ファイルが用意できたら、
partition_criterion_xxx_singlesearch.bat
をテキストエディタで開いて編集します。-fオプションが-f dになっていると思いますが、これを-f Gにします。さらにオプションに-z 比較する系統樹のファイルを加えて下さい。また、-nオプションの文字列も変更して下さい。このオプションは出力ファイルの拡張子を指定するものです。例えば-n calcsitewiseLLとした場合、座位ごとの尤度がRAxML_perSiteLLs.calcsitewiseLLという名前のファイルに保存されます。編集したら念のため別名で保存し、実行して下さい。計算が終わったら、出力されたRAxML_perSiteLLs.calcsitewiseLLのファイル名をRAxML_perSiteLLs.calcsitewiseLL.sitelhに変更して下さい。これは、CONSELが入力ファイルの拡張子を.sitelhと仮定しているためです。
座位ごとの尤度のファイルが用意できたら、CONSELのmakermtコマンドでマルチスケールブートストラップリサンプリングを行います。これは以下のように実行します。
> makermt --puzzle RAxML_perSiteLLs.calcsitewiseLL↓
入力ファイル名には拡張子を付けないことに注意して下さい。マルチスケールブートストラップリサンプリングが正常に終わっていれば、RAxML_perSiteLLs.calcsitewiseLL.rmtというファイルができています。
次に、下記のようにconselコマンドでp値を計算して下さい。
> consel RAxML_perSiteLLs.calcsitewiseLL↓
これでRAxML_perSiteLLs.calcsitewiseLL.pvというファイルができますが、このファイルは人間が見ても意味不明です。これを意味がわかるように表示するのがcatpvです。以下のように実行して下さい。
> catpv RAxML_perSiteLLs.calcsitewiseLL↓
結果は以下のように表示されます。
# reading RAxML_perSiteLLs.calcsitewiseLL.pv # rank item obs au np | bp pp kh sh wkh wsh | # 1 1 -8.4 0.887 0.882 | 0.879 1.000 0.885 0.885 0.885 0.885 | # 2 2 8.4 0.113 0.118 | 0.121 2e-004 0.115 0.115 0.115 0.115 |
rankは尤度による順位、itemは系統樹ファイル中での順序、obsは対数尤度の差、auはAU検定のp値、npはマルチスケールブートストラップリサンプリングから推定された尤度最大となる確率、bpは通常のブートストラップリサンプリングを行なって推定された尤度最大となる確率、ppはベイジアン事後確率、khはKH検定のp値、shはSH検定のp値、wkhはweighted-KH検定のp値、wshはweighted-SH検定のp値となっています。
7.3 MrBayes5Dによる樹形制約付きベイジアン系統樹推定
RAxMLと同様に、TaxonA~TaxonEの5 OTUのデータでTaxonAとTaxonBの単系統性(monophyly)を制約として課す場合を考えましょう。その場合、以下のようなコマンドをNEXUSデータファイル読み込み後に実行することで樹形探索に制約が課されるようになります。コマンドをNEXUSファイルのMrBayesブロックに記述しても結構です(行末にはセミコロンを付加する必要があります)。
MrBayes > Constraint monophyly1 100=TaxonA TaxonB↓ MrBayes > PrSet TopologyPr=Constraints(monophyly1)↓
さらにTaxonAとTaxonBとTaxonCの単系統性も強制する場合は以下のようにします。
MrBayes > Constraint monophyly1 100=TaxonA TaxonB↓ MrBayes > Constraint monophyly2 100=TaxonA TaxonB TaxonC↓ MrBayes > PrSet TopologyPr=Constraints(monophyly1,monophyly2)↓
MrBayes5DもRAxMLと同様に負の制約には対応していません。負の制約を課したい場合には内分枝出現頻度から負の制約を正の制約へ読み替えることで対処する必要があります。
7.4 Bayes factorに基づく仮説比較
複数の系統仮説を比較したいとき、それぞれの仮説を制約として課した解析結果を比較してやることでどちらの仮説が正しいかを検証することができます。ベイズ統計学では、そのような目的にBayes factor (Kass and Raftery, 1995)というものを用います。これは周辺尤度(marginal likelihood)の比に当たります。
多くの分子系統学の論文では、MCMCにおける対数尤度の調和平均(harmonic mean)を周辺尤度の推定値としてBayes factorを算出しますが、この方法ではBayes factorが安定せず、同じ解析を別々に実行してどちらも同じところへ収束していても、どちらか一方を支持してしまう結果を得てしまうことがよくあります。十分に安定したBayes factorを得るには、非常に長いMCMCを走らせなくてはなりません。そこでTracerには、ブートストラップリサンプリングを応用して少ないサンプルからでも高精度にBayes factorを算出する機能が実装されています(Newton and Raftery, 1994)。この機能を用いることで、現実的な計算量でBayes factorを利用した仮説選択が可能です。ここでは、樹形制約1を課したNEXUSファイルconstraint1.nexの解析結果と、樹形制約2を課したNEXUSファイルconstraint2.nexの解析結果を比較する場合を考えます。
MCMCが終わっていれば、constraint1.nex.run1.pとconstraint1.nex.run2.p、constraint2.nex.run1.p、constraint2.nex.run2.pの4つのファイルができているはずです。それぞれのburn-inを決定し(ただしステップ数ではなくサンプル数)、Phylogears2のpgmbburninparamコマンドで2つのburn-in済のログファイルを作成します。それぞれのburn-inを10001、20001、15001、15001、作成するファイルはconstraint1_param.txtとconstraint2_param.txtだとしておくと、コマンドプロンプトかターミナルで以下のようにします。
> pgmbburninparam --burnin=10001 constraint1.nex.run1.p constraint1_param.txt↓ > pgmbburninparam --burnin=20001 --append constraint1.nex.run2.p constraint1_param.txt↓ > pgmbburninparam --burnin=15001 constraint2.nex.run1.p constraint2_param.txt↓ > pgmbburninparam --burnin=15001 --append constraint2.nex.run2.p constraint2_param.txt↓
これで、それぞれの樹形制約を課した解析結果のburn-in済ログファイルが作成できます。
次に、Tracerを起動し、FileメニューのImport Trace File...からconstraint1_param.txtとconstraint2_param.txtを読み込ませます。そして、左上Trace FilesペインでBurn-Inを両方とも0にしてから、両ファイルを選択して反転表示状態にし、AnalysisメニューのCalculate Bayes Factors...からダイアログを呼び出します。ダイアログでは、Likelihood traceをLnLに、Calculate harmonic mean only (no smoothing)のチェックを外し、Bootstrap replicatesを1000以上に設定し、計算を実行します。計算が終わると表が示されるので、Showをln Bayes Factorsに設定します。Trace列が対立仮説のファイル名、対数Bayes factorの値の列名が帰無仮説のファイル名となっています。対数Bayes factorの値から、表7.1の基準で仮説の優劣を判断します(Kass and Raftery, 1995)。
| 対数Bayes factor | 帰無仮説に対して対立仮説が |
|---|---|
| 1~3 | より優れている |
| 3~5 | 強く支持されている |
| 5~ | 非常に強く支持されている |
この方法にも多重比較の問題はあるはずですが、これまでのところそのための補正方法などは普及していません。
前述の通り、MrBayes5Dは2つのMCMCを同時に走らせています。この2つのMCMC間でもBayes factorを算出することができます。もしその2つのMCMCがパラメータ空間上の同じ辺りに収束しているのなら、そのBayes factorによってどちらか一方が支持されることはないはずです。というわけで、「Bayes factorによってどちらか一方への支持が得られてしまう」か否かを収束判定に用いることもできるでしょう。ただ、この方法では「収束していない」ということは分かりますが、「収束している」ということは言えないので注意して下さい。
第8章 参考書籍
最後に、いくつか参考書籍を挙げておきます。
8.1 分子系統学
まず、分子系統学と分子系統解析に関する情報がまとまっている本としては以下の3冊が良いと思います。
分子進化と分子系統学
- 著者
-
根井正利, Sudhir Kumar
- 出版社
-
培風館
- ISBN13
-
978-4563078010
分子進化学を黎明期から支えてこられた根井先生とKumar博士が書かれた本の邦訳です。日本語で分子系統学について幅広く説明されています。分子系統学を体系的に概観するには英語でもこれを上回る本はほとんど無いと思います。
分子系統学への統計的アプローチ-計算分子進化学
- 著者
-
Ziheng Yang
- 出版社
-
共立出版
- ISBN13
-
978-4320056770
分子系統解析法開発の第一人者Yang博士が書かれた本の邦訳。最尤法・ベイズ法や、最先端のトピックスまで扱われた良書です。
Inferring Phylogenies
- 著者
-
Joseph Felsenstein
- 出版社
-
Sinauer Associates Inc.
- ISBN13
-
978-0878931774
分子系統解析に最尤法やブートストラップ法を導入したFelsenstein博士による系統樹推定法を網羅的に解説した決定版的書籍です。
The Phylogenetic Handbook: A Practical Approach to Phylogenetic Analysis and Hypothesis Testing
- 編者
-
Philippe Lemey, Marco Salemi, Anne-Mieke Vandamme
- 出版社
-
Cambridge University Press
- ISBN13
-
978-0521730716
タイトルから分かる通り英語です。とは言え、ソフトウェアの使用方法の解説部分は、使いながら見ればさほど難しいものではないと思います。旧版から大幅に改訂され最新のソフトウェアまでカバーしています。
8.2 統計学
分子系統学は、ある種の「超」応用統計学です。ですから、当然統計学の知識が役に立つ、というか必要になってきます。この本で触れている方法に関連する統計解析法について書かれている本を紹介します。
モデル選択-予測・検定・推定の交差点
- 著者
-
下平英寿, 伊藤 秀一, 久保川達也, 竹内啓
- 出版社
-
岩波書店
- ISBN13
-
978-4000068437
AICの導出過程やKH・SH・AU検定までも説明されています。これらの検定法を使われる方は是非ご一読下さい。
ベイズ統計と統計物理
- 著者
-
伊庭幸人
- 出版社
-
岩波書店
- ISBN13
-
978-4000111584
ベイジアンMCMCについておそらく最も易しく説明されている本です。MrBayesを使いながら読むとパラメータの意味が良く分かるだろうと思います。
計算統計II-マルコフ連鎖モンテカルロ法とその周辺
- 著者
-
伊庭幸人, 種村正美, 大森裕浩, 和合肇, 佐藤整尚, 高橋明彦
- 出版社
-
岩波書店
- ISBN13
-
978-4000068529
ベイジアンMCMCについてもっと深く知りたい方のための本です。
8.3 UNIX入門
分子系統解析を行うソフトウェアは、UNIXの関連知識があると大変楽に使うことができます。以下ではWindows上でUNIXライクな環境を構築できるCygwinの入門書、Linuxの中でも初心者でも比較的取っ付きやすいUbuntu Linuxの入門書、Mac OS XをUNIXとして使うための入門書、シェルの入門書を挙げます。CDやDVDが付属しているものもありますが、この世界は進歩が早いので、ソフトウェアはWebから最新版をダウンロードするようにしましょう。なお、以下の本は必ずしも私は読んではいません。
ちなみに、私が主に使っているUNIXはGentoo Linuxという、マイナーなものです。極限までカスタマイズ・チューニングができるのが特徴です。コンピュータの性能を限界まで引き出したい方は検討されてみるとよいでしょう。公式サイトのハンドブックが大変よくできていますのである程度のUNIX利用経験があれば簡単に使えるようになると思います。
UNIXが使えるようになったら、SSHという遠隔操作するためのソフトウェアと、GNU screenまたはtmuxというソフトを是非インストールしましょう。これらを組み合わせることで、遠隔地からインターネット経由で自宅や研究室の高速なコンピュータに接続して系統解析を行わせ、さらに行わせたままで接続を切ったり再接続したりすることができるようになります。使用方法は、検索すれば説明してくれているWebページがすぐに見つかります。
Windowsで使えるUNIX環境-Cygwin徹底入門
- 著者
-
小川淳一
- 出版社
-
ソーテック社
- ISBN13
-
978-4881663622
WindowsでUNIXを使う本-CygwinでUNIX入門
- 著者
-
阿久津良和
- 出版社
-
毎日コミュニケーションズ
- ISBN13
-
978-4839911959
はじめてのUbuntu-超初心者向けLinuxを使いこなす
- 著者
-
天野友道
- 出版社
-
工学社
- ISBN13
-
978-4777513086
Ubuntuスタートアップバイブル
- 著者
-
佐々木宣文
- 出版社
-
毎日コミュニケーションズ
- ISBN13
-
978-4839930691
Mac OS XユーザのためのUNIX入門-ターミナルから覗くUNIXの世界
- 著者
-
大津真
- 出版社
-
毎日コミュニケーションズ
- ISBN13
-
978-4839909574
入門Unix for Mac OS X
- 著者
-
Dave Taylor
- 出版社
-
オライリージャパン
- ISBN13
-
978-4873112749
シェルの基本テクニック
- 著者
-
西村めぐみ
- 出版社
-
IDGジャパン
- ISBN13
-
978-4872802252
UNIXシェル入門-bashの基本操作とUNIXの環境設定
- 著者
-
北浦訓行, 小島範幸
- 出版社
-
技術評論社
- ISBN13
-
978-4774139203
引用文献
- Ababneh, F., Jermiin, L. S., Ma, C., and Robinson, J., 2006, “Matched-pairs tests of homogeneity with applications to homologous nucleotide sequences”, Bioinformatics, 22, 1225–1231.
- Abascal, F., Posada, D., and Zardoya, R., 2007, “MtArt: a new model of amino acid replacement for Arthropoda”, Molecular Biology and Evolution, 24, 1–5.
- Aberer, A. J., Kobert, K., and Stamatakis, A., 2014, “ExaBayes: massively parallel bayesian tree inference for the whole-genome era”, Molecular Biology and Evolution, 31, No. 10, 2553–2556, Oct.
- Adachi, J. and Hasegawa, M., 1996, “MOLPHY version 2.3: programs for molecular phylogenetics based in maximum likelihood”, Computer Science Monographs, 28, 1–150.
- Adachi, J., Waddell, P. J., Martin, W., and Hasegawa, M., 2000, “Plastid genome phylogeny and a model of amino acid substitution for proteins encoded by chloroplast DNA”, Journal of Molecular Evolution, 50, 348–358.
- Akaike, H., 1974, “New look at statistical-model identification”, IEEE Transactions on Automatic Control, 19, 716–723.
- Altekar, G., Dwarkadas, S., Huelsenbeck, J. P., and Ronquist, F., 2004, “Parallel Metropolis coupled Markov chain Monte Carlo for Bayesian phylogenetic inference”, Bioinformatics, 20, 407–415.
- Avise, J. C. and Robinson, T. J., 2008, “Hemiplasy: a new term in the lexicon of phylogenetics”, Systematic Biology, 57, No. 3, 503–507, Jun.
- Blanquart, S. and Lartillot, N., 2006, “A Bayesian compound stochastic process for modeling nonstationary and nonhomogeneous sequence evolution”, Molecular Biology and Evolution, 23, No. 11, 2058–2071, Nov.
- Blanquart, S. and Lartillot, N., 2008, “A site- and time-heterogeneous model of amino acid replacement”, Molecular Biology and Evolution, 25, No. 5, 842–858, May.
- Boussau, B. and Gouy, M., 2006, “Efficient likelihood computations with nonreversible models of evolution”, Systematic Biology, 55, No. 5, 756–768, Oct.
- Cao, Y., Janke, A., Waddell, P. J., Westerman, M., Takenaka, O., Murata, S., Okada, N., Pääbo, S., and Hasegawa, M., 1998, “Conflict among individual mitochondrial proteins in resolving the phylogeny of eutherian orders.”, Journal of Molecular Evolution, 47, 307–322.
- Capella-Gutiérrez, S., Silla-Martínez, J. M., and Gabaldón, T., 2009, “trimAl: a tool for automated alignment trimming in large-scale phylogenetic analyses”, Bioinformatics, 25, No. 15, 1972–1973, Aug.
- Castresana, J., 2000, “Selection of conserved blocks from multiple alignments for their use in phylogenetic analysis”, Molecular Biology and Evolution, 17, No. 4, 540–552, Apr.
-
Cochran, W. G., 1954, “Some methods for strengthening the common
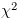 tests”, Biometrics, 10, 417–451.
tests”, Biometrics, 10, 417–451.
- Criscuolo, A. and Gribaldo, S., 2010, “BMGE (Block Mapping and Gathering with Entropy): a new software for selection of phylogenetic informative regions from multiple sequence alignments”, BMC Evolutionary Biology, 10, 210.
- Dayhoff, M. O., Schwartz, R. M., and Orcutt, B. C., 1978, “A model of evolutionary change in proteins, Vol. 5, Suppl. 3”, in Dayhoff, M. O. ed. Atlas of Protein Sequence Structure: National Biomedical Research Foundation, 345–352.
- Dimmic, M. W., Rest, J. S., Mindell, D. P., and Goldstein, R. A., 2002, “rtREV: an amino acid substitution matrix for inference of retrovirus and reverse transcriptase phylogeny”, Journal of Molecular Evolution, 55, 65–73.
- Edgar, R. C., 2004, “MUSCLE: multiple sequence alignment with high accuracy and high throughput”, Nucleic Acids Research, 32, No. 5, 1792–1797.
- Felsenstein, J., 1981, “Evolutionary trees from DNA sequencies - a maximum-likelihood approach”, Journal of Molecular Evolution, 17, 368–376.
- Felsenstein, J., 1985, “Confidence-limits on phylogenies - an approach using the bootstrap”, Evolution, 39, 783–791.
- Fleissner, R., Metzler, D., and von Haeseler, A., 2005, “Simultaneous statistical multiple alignment and phylogeny reconstruction”, Systematic Biology, 54, 548–561.
- Hastings, W. K., 1970, “Monte Carlo sampling methods using Markov chains and their applications”, Biometrika, 57, 97–109.
- Henikoff, S. and Henikoff, J. G., 1992, “Amino acid substitution matrices from protein blocks”, Proceedings of the National Academy of Sciences of the United States of America, 89, 10915–10919.
- Hrdy, I., Hirt, R. P., Dolezal, P., Bardonová, L., Foster, P. G., Tachezy, J., and Embley, T. M., 2004, “Trichomonas hydrogenosomes contain the NADH dehydrogenase module of mitochondrial complex I”, Nature, 432, No. 7017, 618–622, Dec.
- Jobb, G., 2008, “Treefinder version of April 2008”, Software distributed by the author at http://www.treefinder.de/.
- Jobb, G., von Haeseler, A., and Strimmer, K., 2004, “Treefinder: a powerful graphical analysis environment for molecular phylogenetics”, BMC Evolutionary Biology, 4, 18.
- Jones, D. T., Taylor, W. R., and Thornton, J. M., 1992, “The rapid generation of mutation data matrices from protein sequences”, Computer Applications in the Biosciences, 8, 275–282.
- Jukes, T. H. and Cantor, C. R., 1969, “Evolution of protein molecules”, in Munro, H. N. ed. Mammalian protein metabolism, New York: Academic Press, 21–132.
- Kass, R. E. and Raftery, A. E., 1995, “Bayes Factors”, Journal of the American Statistical Association, 90, 773–795.
- Kass, R. E., Carlin, B. P., Gelman, A., and Neal, R., 1998, “Markov chain Monte Carlo in practice: a roundtable discussion”, American Statistician, 52, 93–100.
- Katoh, K., Kuma, K., Toh, H., and Miyata, T., 2005, “MAFFT version 5: improvement in accuracy of multiple sequence alignment”, Nucleic Acids Research, 33, 511–518.
- Kimura, M., 1980, “A simple method for estimating evolutionary rates of base substitutions through comparative studies of nucleotide sequences”, Journal of Molecular Evolution, 16, 111–120.
- Kimura, M., 1983, The neutral theory of molecular evolution: Cambridge University Press.
- Kishino, H. and Hasegawa, M., 1989, “Evaluation of the maximum likelihood estimate of the evolutionary tree topologies from DNA sequence data, and the branching order in hominoidea”, Journal of Molecular Evolution, 29, 170–179.
- Kosiol, C. and Goldman, N., 2005, “Different versions of the Dayhoff rate matrix”, Molecular Biology and Evolution, 22, 193–199.
- Larkin, M. A., Blackshields, G., Brown, N. P., Chenna, R., McGettigan, P. A., McWilliam, H., Valentin, F., Wallace, I. M., Wilm, A., Lopez, R., Thompson, J. D., Gibson, T. J., and Higgins, D. G., 2007, “Clustal W and Clustal X version 2.0”, Bioinformatics, 23, No. 21, 2947–2948, Nov.
- Lartillot, N. and Philippe, H., 2004, “A Bayesian mixture model for across-site heterogeneities in the amino-acid replacement process”, Molecular Biology and Evolution, 21, 1095–1109.
- Le, S. Q. and Gascuel, O., 2008, “An improved general amino acid replacement matrix”, Molecular Biology and Evolution, 25, 1307–1320.
- Le, S. Q. and Gascuel, O., 2010, “Accounting for solvent accessibility and secondary structure in protein phylogenetics is clearly beneficial”, Systematic Biology, 59, No. 3, 277–287.
- Le, S. Q., Dang, C. C., and Gascuel, O., 2012, “Modeling protein evolution with several amino acid replacement matrices depending on site rates”, Molecular Biology and Evolution, 29, No. 10, 2921–2936, Oct.
- Lunter, G., Miklós, I., Drummond, A., Jensen, J. L., and Hein, J., 2005, “Bayesian coestimation of phylogeny and sequence alignment”, BMC Bioinformatics, 6, 83.
- Metropolis, N., Rosenbluth, A. W., Rosenbluth, M. N., and Teller, A. H., 1953, “Equation of state calculations by fast computing machines”, Journal of Chemical Physics, 21, 1087–1092.
- Misof, B. and Misof, K., 2009, “A Monte Carlo approach successfully identifies randomness in multiple sequence alignments: a more objective means of data exclusion”, Systematic Biology, 58, No. 1, 21–34, Feb.
- Müller, T. and Vingron, M., 2000, “Modeling amino acid replacement”, Journal of Computational Biology, 7, 761–776.
- Newton, M. A. and Raftery, A. E., 1994, “Approximate Bayesian inference with the weighted likelihood bootstrap”, Journal of the Royal Statistical Society, 56, 3–48.
- Nickle, D. C., Heath, L., Jensen, M. A., Gilbert, P. B., Mullins, J. I., and Pond, S. L. K., 2007, “HIV-specific probabilistic models of protein evolution”, PLoS ONE, 2, e503.
- Pagel, M. and Meade, A., 2004, “A phylogenetic mixture model for detecting pattern-heterogeneity in gene sequence or character-state data”, Systematic Biology, 53, 571–581.
- Posada, D. and Crandall, K. A., 1998, “Modeltest: testing the model of DNA substitution”, Bioinformatics, 14, 817–818.
- Redelings, B. D. and Suchard, M. A., 2005, “Joint Bayesian estimation of alignment and phylogeny”, Systematic Biology, 54, 401–418.
- Ronquist, F. and Huelsenbeck, J. P., 2003, “MrBayes 3: Bayesian phylogenetic inference under mixed models”, Bioinformatics, 19, 1572–1574.
- Ronquist, F., Huelsenbeck, J. P., and van der Mark, P., 2005, “MrBayes 3.1 Manual 5/26/2005”, Distributed at http://mrbayes.csit.fsu.edu/manual.php.
- Rota-Stabelli, O., Yang, Z., and Telford, M. J., 2009, “MtZoa: a general mitochondrial amino acid substitutions model for animal evolutionary studies”, Molecular Phylogenetics and Evolution, 52, No. 1, 268–272, Jul.
- Saitou, N. and Nei, M., 1987, “The neighbor-joining method: a new method for reconstructing phylogenetics trees”, Molecular Biology and Evolution, 4, 406–425.
- Schwarz, G., 1978, “Estimating the dimension of a model”, Annals of Statistics, 6, 461–464.
- Shimodaira, H., 2002, “An approximately unbiased test of phylogenetic tree selection”, Systematic Biology, 51, 492–508.
- Shimodaira, H. and Hasegawa, M., 1999, “Multiple comparisons of log-likelihoods with applications to phylogenetic inference”, Molecular Biology and Evolution, 16, 1114–1116.
- Stamatakis, A., 2006, “RAxML-VI-HPC: maximum likelihood-based phylogenetic analyses with thousands of taxa and mixed models”, Bioinformatics, 22, 2688–2690.
- Sugiura, N., 1978, “Further analysis of the data by Akaike’s information criterion and the finite corrections”, Communications in Statistics: Theory and Methods, A7, 13–26.
- Swofford, D. L., 2003, PAUP*. Phylogenetic Analysis Using Parsimony (*and Other Methods). Version 4, Sunderland, Massachusetts: Sinauer Associates.
- Swofford, D. L. and Begle, D. P., 1993, PAUP: Phylogenetic Analysis Using Parsimony , Ver.3.1. User’s Manual: Laboratory of Molecular Systematics, Smithonian Institution.
- Talavera, G. and Castresana, J., 2007, “Improvement of phylogenies after removing divergent and ambiguously aligned blocks from protein sequence alignments”, Systematic Biology, 56, No. 4, 564–577, Aug.
- Tanabe, A. S., 2011, “Kakusan4 and Aminosan: two programs for comparing nonpartitioned, proportional and separate models for combined molecular phylogenetic analyses of multilocus sequence data”, Molecular Ecology Resources, 11, No. 5, 914–921, Sep.
- Tavaré, S., 1986, “Some probabilistic and statistical problems in the analysis of DNA sequences”, Lectures on Mathematics in the Life Sciences, 17, 57–86.
- Tuffley, C. and Steel, M., 1997, “Links between maximum likelihood and maximum parsimony under a simple model of site substitution”, Bulletin of Mathematical Biolology, 59, No. 3, 581–607, May.
- Tuffley, C. and Steel, M., 1998, “Modeling the covarion hypothesis of nucleotide substitution”, Mathematical Biosciences, 147, No. 1, 63–91, Jan.
- Veerassamy, S., Smith, A., and Tillier, E. R. M., 2003, “A transition probability model for amino acid substitutions from blocks.”, Journal of Computational Biology, 10, 997–1010.
- Venditti, C., Meade, A., and Pagel, M., 2006, “Detecting the node-density artifact in phylogeny reconstruction”, Systematic Biology, 55, No. 4, 637–643, Aug.
- Webster, A. J., Payne, R. J. H., and Pagel, M., 2003, “Molecular phylogenies link rates of evolution and speciation”, Science, 301, No. 5632, 478, Jul.
- Whelan, S. and Goldman, N., 2001, “A general empirical model of protein evolution derived from multiple protein families using a maximum-likelihood approach”, Molecular Biology and Evolution, 18, 691–699.
- Woese, C. R., Achenbach, L., Rouviere, P., and Mandelco, L., 1991, “Archaeal phylogeny: reexamination of the phylogenetic position of Archaeoglobus fulgidus in light of certain composition-induced artifacts”, Systematic and Applied Microbiology, 14, No. 4, 364–371.
- Yang, Z., 1993, “Maximum likelihood estimation of phylogeny from DNA sequences when substitution rates differ over sites”, Molecular Biology and Evolution, 10, 1396–1401.
- Yang, Z., 1994, “Maximum likelihood phylogenetic estimation from DNA sequences with variable rates over sites: approximate methods”, Journal of Molecular Evolution, 39, 306–314.
- Yang, Z., 1995, “A space-time process model for the evolution of DNA sequences”, Genetics, 139, 993–1005.